Annonce importante
L’amylose est un groupe de maladies qui partagent une caractéristique commune du dépôt de fibrilles amyloïdes dans divers organes et tissus. Les types d’amyloïdose systémique sont tous très différents les uns des autres en ce qui concerne la nature biochimique des dépôts amyloïdes et de la protéine amyloïde précurseur. Certains sont acquis et d’autres sont hérités. Certains types partagent les caractéristiques cliniques de la maladie en ce qui concerne l’atteinte et le dysfonctionnement des organes. Il est crucial de définir la protéine amyloïdogène précurseur et de dactylographier avec précision les dépôts amyloïdes pour pouvoir offrir un traitement précis, évaluer le pronostic et suggérer un conseil génétique au besoin.
amylose AL (primaire)
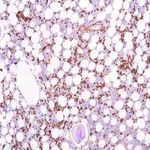
L’amylose AL (chaîne légère d’immunoglobuline) est un trouble plasmatique acquis dans lequel une chaîne légère d’immunoglobuline monoclonale est produite dans la moelle osseuse et se trouve généralement dans le sang ou l’urine. L’amylose AL survient occasionnellement avec un myélome multiple, un lymphome ou une macroglobulinémie de Waldenstorm. Les fibrilles amyloïdes dans ce type d’amylose sont constituées de protéines de la chaîne légère des immunoglobulines (kappa ou lambda). Les symptômes peuvent survenir dans n’importe quel organe du corps et comprennent une insuffisance cardiaque, des protéines dans l’urine ou une insuffisance rénale, une hypertrophie du foie, une neuropathie ou une hypertrophie de la langue. Un traitement par chimiothérapie, y compris une chimiothérapie à forte dose avec transplantation de cellules souches et d’autres agents, peut mettre la maladie de la moelle osseuse en rémission, entraînant une amélioration du dysfonctionnement des organes et protégeant les organes cibles. C’est l’un des types d’amylose les plus courants diagnostiqués aux États-Unis.
amylose AA (secondaire)
L’amylose AA est causée par une infection chronique ou une maladie inflammatoire telle que la polyarthrite rhumatoïde, la fièvre méditerranéenne familiale (FMF), l’ostéomyélite ou l’iléite granulomateuse. L’infection ou l’inflammation provoque une élévation d’une protéine de phase aiguë, la SAA, dont une partie se dépose sous forme de fibrilles amyloïdes. Par conséquent, on l’appelle amylose AA. L’amylose AA commence généralement par une maladie des reins, mais d’autres organes peuvent être affectés. Le traitement médical ou chirurgical de l’infection chronique ou de la maladie inflammatoire sous-jacente peut ralentir ou arrêter la progression de ce type d’amylose.
ATTR amyloidosis
ATTR amyloidosis (ATTRm or ATTRwt)
There are several types of inherited amyloidoses, the most common of which is caused by a mutation in the transthyretin (TTR) gene that produces abnormal transthyretin protein. The abnormal TTR protein deposits as amyloid fibrils: Hence, it is termed ATTR amyloidosis. Les symptômes de la maladie sont généralement une neuropathie et une cardiomyopathie et surviennent au milieu ou à la fin de la vie. L’amylose ATTR se rencontre dans les familles de presque toutes les origines ethniques. Plus de 100 mutations différentes de la transthyrétine sont connues et la plupart provoquent une amylose. L’ATTR peut également se produire avec la protéine TTR non mutée de type sauvage, provoquant généralement une cardiomyopathie chez les hommes plus âgés; auparavant appelée « amylose systémique sénile”, elle est maintenant connue sous le nom d’amylose ATTRwt. Les options de traitement incluent des stabilisants TTR (Diflunisal ou Tafamidis), ou des silencieux géniques ou la production de TTR ou la transplantation hépatique.
Autres amyloïdoses héréditaires
Il existe d’autres mutations génétiques qui produisent des protéines responsables de l’amylose. Ce sont très rares. Ils comprennent l’apolipoprotéine A-I (AApoAI), l’apolipoprotéine A-II (AApoAII), la gelsoline (AGel), le fibrinogène (AFib) et le lysozyme (ALys). Une protéine connue sous le nom de Lect2 peut également provoquer une amylose; on ne sait pas si cela est héréditaire ou non.
L’amylose à la microglobuline bêta-2 (Abeta2m)
L’amylose à la microglobuline bêta-2 est causée par une insuffisance rénale chronique et survient souvent chez les patients dialysés depuis de nombreuses années. Les dépôts amyloïdes sont constitués de la protéine bêta-2 microglobuline qui s’est accumulée dans les tissus, en particulier autour des articulations, lorsqu’elle ne peut pas être excrétée par les reins en raison d’une insuffisance rénale.
amylose localisée (ALoc)
Il existe de nombreux types d’amyloïdoses localisées. Les dépôts amyloïdes localisés dans les voies respiratoires (trachée ou bronche), les yeux ou la vessie sont souvent causés par la production locale de chaînes légères d’immunoglobulines, qui ne proviennent pas de la moelle osseuse. L’AL localisée peut être traitée par radiothérapie, et elle et d’autres formes localisées peuvent parfois être chirurgicales. D’autres types localisés d’amylose sont associés à des protéines endocrines, ou à des protéines produites dans la peau, le cœur et d’autres sites. Ceux-ci ne deviennent généralement pas systémiques.