La leucémie à lymphocytes granulaires de grande taille comme complication de la polyarthrite rhumatoïde / Reumatología Clínica
Introduction
Le syndrome de Felty (FS) est une complication systémique rare (moins de 1%) de la polyarthrite rhumatoïde (PR), caractérisée par la triade de la PR, de la neutropénie persistante (3
) et de la splénomégalie de taille variable qui peut aller de la splénomégalie subclinique, uniquement détectable par imagerie, à la splénomégalie massive1. Il survient principalement dans les cas de longue date présentant une maladie articulaire grave et des manifestations extra-articulaires, et a une forte association avec l’haplotype HLA-DR4 (près de 95% des cas).2 Chez 30% à 40% des patients atteints de FS, il y a une expansion des grands lymphocytes granulaires (LGL).1 LGL représentent 10% à 15% des cellules mononucléées circulantes et sont morphologiquement identifiées par leur grande taille (15–18µm), leur noyau rond ou en retrait et leur cytoplasme abondant avec des granules azurophiles. Le phénotype de ces cellules peut être un lymphocyte T cytotoxique (CD8+, CD57+) ou un tueur naturel (NK) (CD3−, CD8−, CD56+).3 Lorsque l’expansion de la LGL est monoclonale et est associée à une infiltration de la moelle osseuse et de la rate par ces cellules, elle est appelée leucémie à grands lymphocytes granulaires (LGLL) et est considérée comme un trouble lymphoprolifératif chronique de bas grade. Sa présentation clinique est similaire à celle de la FS, mettant en évidence la susceptibilité accrue aux infections bactériennes associées à la neutropénie, à l’anémie et à la splénomégalie, comme on l’a également appelé « pseudo-Felty”.3,4 Présentation clinique
À l’âge de 43 ans, un travailleur retraité d’une carrière de granit âgé de 70 ans a reçu un diagnostic de PR séropositive avec atteinte des mains, des pieds, des genoux et des hanches. Il a ensuite développé une pneumoconiose et des nodules rhumatoïdes pulmonaires (Fig. 1), étant diagnostiqué comme le syndrome de Caplan. Au cours de sa progression, il a été traité avec des AINS, des corticostéroïdes, des sels d’or, de la cyclosporine et du méthotrexate. Malgré cela, le patient a développé des dommages structurels aux mains, aux pieds et aux hanches, nécessitant la pose d’une prothèse dans les deux hanches à 51 et 54 ans, respectivement. Ces dernières années, sa maladie est restée stable, traitée avec du méthotrexate 10 mg par semaine et de faibles doses de glucocorticoïdes, sans aucune preuve d’activité articulaire inflammatoire. Il a présenté des déformations en « col de cygne » dans tous les doigts et des nodules rhumatoïdes sur les coudes. il y a 12 mois, il a soudainement développé de la fièvre et des douleurs à l’aine droite, avec choc septique subséquent, démontrant une infection de la hanche droite par Salmonella spp. Le méthotrexate et les glucocorticoïdes ont été suspendus et il a été traité par une antibiothérapie prolongée et un remplacement partiel de la prothèse. Les tests de laboratoire ont mis en évidence une neutropénie persistante, malgré le retrait des médicaments myélotoxiques et l’amélioration de la septicémie, atteignant 0neutrophiles / mm3. Rétrospectivement, lors de l’examen du nombre de neutrophiles, il avait de faibles dénombrements un an plus tôt, entre 1800 et 1000 / mm3. Le reste de la numération formule sanguine et de la biochimie était normal. La VS était de 80 mm / h et la CRP de 111 mg / l. Il maintenait des niveaux élevés de facteur rhumatoïde (6930U / ml) et d’anti-CCP (300U / ml) et présentait également une hypergammaglobulinémie polyclonale. Les anticorps antinucléaires et les antigènes nucléaires extractibles étaient négatifs et les niveaux de complément se situaient dans les valeurs normales. Le typage HLA a montré qu’il portait l’haplotype DRB1 * 0404 (DR4) et une splénomégalie légère a été détectée (13,7 cm) par tomodensitométrie abdominale.

Pneumoconiose et nodules rhumatoïdes pulmonaires.
Le frottis de sang périphérique a montré une lymphocytose par LGL (Fig. 2), qui dans l’immunophénotype correspondait à 42% des leucocytes totaux, avec un phénotype aberrant de lymphocyte T cytotoxique (CD3 +, CD8 +, CD5 +, CD7 + /-, CD4-, CD56- et DR +). La biopsie de moelle osseuse a montré 20% de la cellularité totale de la moelle correspondant à la même expansion clonale (confirmée par réarrangement de la région variable du gamma TCR). Sur la base de ces résultats, un diagnostic de LGLL a été posé et le traitement par méthotrexate 15 mg par semaine a repris malgré 3 mois de neutropénie persistante (3
), nécessitant une administration fréquente de facteurs stimulateurs de la granulopoïèse. Il a ensuite été traité avec du cyclophosphamide, de la vincristine et de la prednisone à fortes doses. Après 6 mois de traitement, la neutropénie persiste.
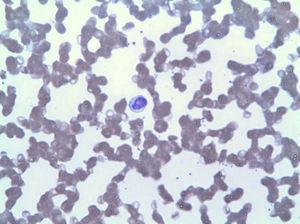
Lymphocytose due à la LGL.
Discussion
La LLL est une leucémie chronique caractérisée par une expansion du phénotype monoclonal LGL des lymphocytes T cytotoxiques activés ou moins fréquemment des cellules NK.5 L’âge moyen du diagnostic est de 60 ans et il est fréquemment associé à des maladies auto-immunes, en particulier la PR, mais a également été décrit dans la colite ulcéreuse, le syndrome de Sjögren, le lupus érythémateux et la sclérose multiple1. Les patients atteints de PR qui ont une LGLL associée ont une présentation clinique similaire à celle de la FS. Il s’agit généralement de patients présentant une PR de longue date, des lésions articulaires graves et des conséquences importantes, ainsi qu’une fréquence accrue de manifestations extra-articulaires telles que nodules rhumatoïdes, adénopathie, ulcères prétibiaux, pleurite, pigmentation de la peau, neuropathie ou épisclérite.6 Le patient présentait également une autre complication rare de la PR, de la pneumoconiose rhumatoïde ou du syndrome de Caplan, caractérisée par l’apparition de nodules pulmonaires avec une histopathologie similaire à celle des nodules rhumatoïdes typiques chez les patients ayant des antécédents d’exposition professionnelle à des poussières inorganiques telles que la silice, le charbon ou le granit.7 A notre connaissance, le cas présent est le premier décrivant la présentation du syndrome de Caplan et de la LLL chez le même patient. Dans la plupart des cas, la LLL de présentation est une neutropénie sévère associée à des infections bactériennes récurrentes. Les microorganismes les plus couramment impliqués sont Staphylococcus aureus, Streptococcus spp. et des bacilles à gram négatif. Plus rarement, une anémie, de la fièvre, des sueurs nocturnes et un élargissement du foie et de la rate 5 peuvent également l’accompagner. Jusqu’à un tiers des patients atteints de LLGG n’ont aucune activité clinique apparente de PR au moment du diagnostic, mais maintiennent des niveaux élevés de VS.6 Jusqu’à 40% des patients atteints de FS ont une lymphocytose LGL.7 Ce fait, ainsi que la similitude clinique et l’association avec HLA-DR4 ont fortement suggéré que FS et LGLL associées à la PR sont des expressions d’une même entité caractérisée par la prolifération de LGL.8 D’autres formes comprennent également des formes plus douces telles que la lymphocytose réactive et les infections à des formes plus agressives de NK5 LGLL. Le diagnostic de LGLL repose sur la découverte d’une expansion monoclonale de LGL dans le sang périphérique et la moelle osseuse avec un immunophénotype caractéristique (CD3 +, CD4−, CD8 +, CD16 +, CD28− et CD57 +). La clonalité est confirmée par l’étude du gène reTCR9. En général, le LGL a une progression chronique et indolente, avec une survie moyenne de 10 ans.1 Dans de rares cas, en particulier lorsque l’expansion est due à LGL avec phénotype NK, cette leucémie peut se comporter de manière plus agressive.5 Les indications les plus courantes pour le traitement sont les infections récurrentes et, moins fréquemment, l’anémie, la splénomégalie symptomatique ou l’apparition de symptômes B sévères B1.
Les traitements de première intention dans les LLL sont des médicaments immunosuppresseurs seuls, en particulier le méthotrexate (10 mg / semaine), la cyclosporine A (1 à 1,5 mg / kg / 2 fois par jour) ou le cyclophosphamide oral (50 à 100 mg / jour). Ce traitement est efficace chez environ 50% des patients, permettant la correction des cytopénies, mais n’éradiquant pas les cellules leucémiques.1 Les glucocorticoïdes peuvent être utilisés pour accélérer la réponse et les facteurs stimulants de la granulopoïèse sont utiles dans la prise en charge initiale de la neutropénie. Chez les patients réfractaires et chez ceux avec un traitement de présentation très agressif avec des schémas chimiothérapeutiques similaires à CHOP (cyclophosphamide, vincristine, doxorubicine et prednisone) et d’autres schémas de lymphome ont été essayés, mais n’ont pas clairement démontré leur efficacité. Les autres traitements testés sont les analogues de la purine, l’Alemtuzumab, le bortézomib, la splénectomie et la greffe de moelle osseuse allogénique, avec des résultats variables.3
Conclusions
La FS et la LLL sont des complications rares de la PR, qui apparaissent dans une maladie de longue date, avec des dommages structurels importants et des manifestations extra-articulaires. Chez les patients atteints de PR de longue date et de neutropénie, la présence de proliférations clonales de LGL dans le sang périphérique et / ou la moelle osseuse doit être exclue, ce qui permet le diagnostic de LGLL. Le traitement de première intention consiste à utiliser des médicaments immunosuppresseurs, tels que le méthotrexate à faible dose, et les glucocorticoïdes peuvent être associés à des facteurs stimulants de la granulopoïèse. D’autres modalités de traitement telles que la chimiothérapie ou la splénectomie ont montré des résultats variables dans certains cas réfractaires.
Divulgations éthiques
Protection des sujets humains et animaux. Les auteurs déclarent qu’aucune expérience n’a été réalisée sur des humains ou des animaux pour cette enquête.
Confidentialité des données. Les auteurs déclarent avoir suivi les protocoles de leur centre de travail sur la publication des données des patients et que tous les patients inclus dans l’étude ont reçu suffisamment d’informations et ont donné leur consentement éclairé par écrit pour participer à cette étude.