Nagy szemcsés limfocita leukémia, mint a rheumatoid Arthritis szövődménye/Reumatologus a Cl-ben a Cl-ben a
Bevezetés a Felty-szindróma (FS) a rheumatoid arthritis (RA) ritka szisztémás szövődménye (kevesebb, mint 1%), amelyet az RA hármasa, a perzisztens neutropenia (3
) és a változó méretű splenomegalia jellemez, amely a szubklinikai splenomegáliától, csak képalkotással kimutatható, a massive1 splenomegáliáig terjedhet. Ez akkor fordul elő elsősorban a hosszú távú esetekben súlyos ízületi betegség és extraartikuláris megnyilvánulásai, és van egy erős kapcsolat a HLA-DR4 haplotípus (közel 95%-ában).2 Az FS-ben szenvedő betegek 30-40% – ában nagy szemcsés limfociták (LGL) bővülnek.1 az LGL a keringő mononukleáris sejtek 10-15% – át teszi ki, és morfológiailag nagy méretük (15-18), kerek vagy behúzott magjuk és bőséges citoplazma alapján azonosítják őket azurofil granulátumokkal. Ezeknek a sejteknek a fenotípusa lehet citotoxikus T− limfocita (CD8+, CD57+) vagy természetes gyilkos (nk) (CD3−, CD8 -, CD56+).3 Ha az LGL expanziója monoklonális, és ezen sejtek által a csontvelő és a lép infiltrációjával jár, akkor nagy szemcsés limfocita leukémiának (LGLL) nevezik, és krónikus, alacsony fokú limfoproliferatív rendellenességnek tekintik. Klinikai megjelenése hasonló az FS-hez, kiemelve a neutropeniával, vérszegénységgel és splenomegáliával járó bakteriális fertőzések iránti fokozott érzékenységet, amint azt “pszeudo-Felty” – nek is nevezik.3,4 klinikai megjelenés
egy gránitbányából származó 70 éves nyugdíjas munkavállalót 43 éves korában szeropozitív RA-val diagnosztizáltak, kéz, láb, térd és csípő bevonásával. Ezt követően pneumoconiosis és pulmonalis rheumatoid csomók alakultak ki nála. 1), Caplan-szindrómaként diagnosztizálták. Progressziója során NSAID-okkal, kortikoszteroidokkal, aranysókkal, ciklosporinnal és metotrexáttal kezelték. Ennek ellenére a beteg szerkezeti károsodást okozott a kezekben, a lábakban és a csípőben, ami 51, illetve 54 éves korban szükségessé tette a protézis elhelyezését mindkét csípőben. Az utóbbi években betegsége stabil maradt, hetente 10 mg metotrexáttal és alacsony dózisú glükokortikoidokkal kezelték, gyulladásos ízületi aktivitás nélkül. Bemutatta az összes ujj libanyak deformációját és a könyök reumatoid csomóit. 12 hónappal ezelőtt hirtelen láz és fájdalom alakult ki a jobb ágyékában, ezt követően szeptikus sokk, amely a jobb csípő fertőzését mutatta Salmonella spp. A metotrexátot és a glükokortikoidokat felfüggesztették, és hosszan tartó antibiotikum-terápiával és a protézis részleges pótlásával kezelték. A laboratóriumi vizsgálatok tartós neutropeniát igazoltak a myelotoxikus gyógyszerek visszavonása és a szepszis javulása ellenére, elérve a 0neutrophil/mm3 értéket. Visszamenőleg, a neutrofilek számának áttekintésekor egy évvel korábban alacsony volt a száma, 1800-1000 / mm3 között. A vérkép és a biokémia többi része normális volt. Az ESR 80mm/h és CRP 111MG/l volt. magas szintű rheumatoid faktort (6930U/ml) és anti-CCP-t (300U / ml) tartott fenn, és poliklonális hipergammaglobulinémiája is volt. Az antinukleáris antitestek és az extrahálható nukleáris antigének negatívak voltak, a komplementszintek pedig a normál értéken belül voltak. A HLA tipizálás azt mutatta, hogy a DRB1*0404 (DR4) haplotípust hordozta, és enyhe splenomegáliát (13,7 cm) mutattak ki hasi számítógépes tomográfiával.

Pneumoconiosis és pulmonalis rheumatoid csomók.
a perifériás vérkenet LGL által mutatott limfocitózist mutatott (ábra. 2), amely az immunfenotípusban az összes leukocita 42%− ának felel meg, a citotoxikus T− limfocita aberráns fenotípusával (CD3+, CD8+, CD5+, CD7+/−, CD4 -, CD56-és DR+). A csontvelő biopszia a teljes csontvelő-cellularitás 20% – át mutatta, amely azonos klonális expanziónak felel meg (a TCR gamma változó régiójának átrendeződésével megerősítve). Ezen eredmények alapján diagnosztizálták az LGLL-t, és a heti 15 mg metotrexát-kezelést 3 hónap ellenére újrakezdték, tartós neutropeniával (3), amely granulopoiesist stimuláló faktorok gyakori alkalmazását tette szükségessé. Ezt követően nagy dózisú ciklofoszfamiddal, vinkrisztinnel és prednizonnal kezelték. 6 hónapos kezelés után a neutropenia továbbra is fennáll.
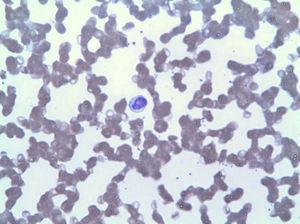
Lymphocytosis az LGL miatt.
Vita
az LGLL egy krónikus leukémia, amelyet az aktivált citotoxikus T-limfociták vagy ritkábban az NK-sejtek LGL monoklonális fenotípusának terjeszkedése jellemez.5 a diagnózis átlagos életkora 60 év, és gyakran társul autoimmun betegségekkel, különösen az RA-val, de leírták a colitis ulcerosa, az SJ XXL-Gren-szindróma, a lupus erythematosus és a multiple1 sclerosis esetében is. Az RA-ban szenvedő betegek, akik az LGLL-t társították, az FS-hez hasonló klinikai megjelenéssel rendelkeznek. Általában régóta fennálló RA-val, súlyos ízületi károsodással és jelentős következményekkel, valamint az extraartikuláris megnyilvánulások, például rheumatoid csomók, lymphadenopathia, pretibialis fekélyek, pleuritis, bőr pigmentáció, neuropathia vagy episcleritis fokozott gyakoriságával rendelkeznek.6 a betegnek volt egy másik ritka szövődménye az RA – nak, a rheumatoid pneumoconiosisnak vagy a Caplan-szindrómának, amelyet a tipikus reumatoid csomókhoz hasonló hisztopatológiájú pulmonalis csomók megjelenése jellemez azoknál a betegeknél, akiknek kórtörténetében szervetlen porok, például szilícium-dioxid, szén vagy gránit foglalkozási expozíciója volt.7 tudomásunk szerint a jelen eset az első, amely a Caplan-szindróma és az LGLL megjelenését írja le ugyanabban a betegben. A legtöbb esetben a bemutató LGLL súlyos neutropenia, amely visszatérő bakteriális fertőzésekkel jár. A leggyakrabban érintett mikroorganizmusok a Staphylococcus aureus, a Streptococcus spp. és gram-negatív bacillusok. Ritkábban vérszegénység, láz, éjszakai izzadás és máj-és lépnagyobbítás5 is kísérheti. Az LLGG-ben szenvedő betegek legfeljebb egyharmadának nincs nyilvánvaló RA klinikai aktivitása a diagnózis idején, de fenntartja az ESR magas szintjét.6 Az FS-ben szenvedő betegek legfeljebb 40% – ánál van LGL limfocitózis.7 Ez a tény, a klinikai hasonlósággal és a HLA-DR4-gyel való összefüggéssel együtt erősen azt sugallja, hogy az RA-val társított FS és LGLL ugyanazon entitás kifejeződései, amelyeket az LGL proliferációja jellemez.8 egyéb formák közé tartoznak az enyhébb formák is, mint például a reaktív limfocitózis és az NK5 LGLL agresszívebb formáinak fertőzései. Az LGLL diagnózisa az LGL perifériás vérben és csontvelőben történő monoklonális expanziójának megállapításán alapul, jellegzetes immunfenotípussal (CD3+, CD4−, CD8+, CD16+, CD28− és CD57+). A klónozást a reTCR9 gén tanulmányozása igazolja. Általában az LGL krónikus és indolens progresszióval rendelkezik, átlagos túlélése 10 év.1 ritka esetekben, különösen akkor, ha a terjeszkedés az NK fenotípusú LGL-nek köszönhető, ez a leukémia agresszívebben viselkedhet.5 a kezelés leggyakoribb indikációja a visszatérő fertőzések, ritkábban a vérszegénység, a tüneti splenomegalia vagy a súlyos B tünetek megjelenése B1.
az LGLL első vonalbeli kezelése önmagában immunszuppresszív gyógyszerek, konkrétan metotrexát (10 mg/hét), ciklosporin A (1-1, 5 mg/kg/2–szer naponta) vagy orális ciklofoszfamid (50-100mg/nap). Ez a kezelés a betegek mintegy 50% – ánál hatékony, elérve a citopeniák korrekcióját, de nem szünteti meg a leukémiás sejteket.1 glükokortikoidok alkalmazhatók a reakció felgyorsítására, és a granulopoiesist stimuláló faktorok hasznosak a neutropenia kezdeti kezelésében. Refrakter betegeknél és azoknál, akiknek nagyon agresszív a prezentációja, a Chop-hoz hasonló kemoterápiás kezeléssel (ciklofoszfamid, vinkrisztin, doxorubicin és prednizon) és más lymphoma-kezelési sémákkal próbálkoztak, de ezek hatékonyságát nem bizonyították egyértelműen. Egyéb tesztelt kezelések a purin analógok, az Alemtuzumab, a bortezomib, a splenectomia és az allogén csontvelő-transzplantáció, változó eredménnyel.3
következtetések
mind az FS, mind az LGLL az RA ritka szövődményei, amelyek hosszú távú betegségben jelentkeznek, jelentős szerkezeti károsodással és extraartikuláris megnyilvánulásokkal. Régóta fennálló RA és neutropenia esetén ki kell zárni az LGL klonális proliferációjának jelenlétét a perifériás vérben és/vagy a csontvelőben, lehetővé téve az LGLL diagnosztizálását. Az első vonalbeli kezelés immunszuppresszív gyógyszerek, például alacsony dózisú metotrexát alkalmazása, a glükokortikoidok pedig granulopoiesist stimuláló faktorokkal társulhatnak. Más kezelési módok, például kemoterápia vagy splenectomia változó eredményeket mutattak néhány refrakter esetben.
etikai közlemények
emberi és állati alanyok védelme. A szerzők kijelentik, hogy e vizsgálat során nem végeztek kísérleteket embereken vagy állatokon.
Az adatok titkossága. A szerzők kijelentik, hogy betartották a munkacsoportjuknak a betegadatok közzétételére vonatkozó protokolljait, és hogy a vizsgálatban részt vevő valamennyi beteg elegendő információt kapott, és írásban tájékozott beleegyezését adta a vizsgálatban való részvételhez.