A grandi Linfociti Granulari Leucemia come Complicanza di Artrite Reumatoide | Reumatología Clinica
Introduzione
Feltrosa sindrome di (FS) è un raro sistemica complicazione (meno dell ‘ 1%) di artrite reumatoide (RA), caratterizzata dalla triade di RA, neutropenia persistente (3
) e splenomegalia di varie dimensioni che possono variare da subclinica splenomegalia, rilevabili solo mediante imaging, per massive1 splenomegalia. Si verifica principalmente in casi di lunga data con grave malattia articolare e manifestazioni extraarticolari e ha una forte associazione con l’aplotipo HLA-DR4 (quasi il 95% dei casi).2 Nel 30% -40% dei pazienti con FS c’è un’espansione di grandi linfociti granulari (LGL).1 LGL rappresentano il 10% -15% delle cellule mononucleate circolanti e sono morfologicamente identificati dalle loro grandi dimensioni (15–18µm), dal loro nucleo rotondo o dentellato e dall’abbondante citoplasma con granuli azurofili. Il fenotipo di queste cellule può essere citotossico linfociti T (CD8+, CD57+) o natural killer (NK) (CD3−, CD8−, CD56+).3 Quando l’espansione di LGL è monoclonale ed è associata all’infiltrazione del midollo osseo e della milza da parte di queste cellule, si chiama grande leucemia linfocitaria granulare (LGLL) ed è considerata una malattia linfoproliferativa cronica di basso grado. La sua presentazione clinica è simile a quella di FS, evidenziando l’aumentata suscettibilità alle infezioni batteriche associate a neutropenia, anemia e splenomegalia, come è stato anche chiamato “pseudo-Felty”.3,4 Presentazione clinica
Un lavoratore pensionato di 70 anni di una cava di granito è stato diagnosticato all’età di 43 anni con AR sieropositivo con coinvolgimento di mani, piedi, ginocchia e fianchi. Successivamente ha sviluppato pneumoconiosi e noduli reumatoidi polmonari (Fig. 1), essendo diagnosticata come sindrome di Caplan. Durante la sua progressione, è stato trattato con FANS, corticosteroidi, sali d’oro, ciclosporina e metotrexato. Nonostante ciò, il paziente ha sviluppato danni strutturali alle mani, ai piedi e ai fianchi, richiedendo il posizionamento della protesi in entrambi i fianchi rispettivamente a 51 e 54 anni. Negli ultimi anni, la sua malattia è rimasta stabile, trattata con metotrexato 10 mg settimanali e basse dosi di glucocorticoidi, senza alcuna evidenza di attività infiammatoria articolare. Ha presentato deformità a collo d’oca in tutte le dita e noduli reumatoidi sui gomiti. 12 mesi fa ha improvvisamente sviluppato febbre e dolore all’inguine destro, con conseguente shock settico, dimostrando un’infezione dell’anca destra da Salmonella spp. Metotrexato e glucocorticoidi sono stati sospesi e sono stati trattati con terapia antibiotica prolungata e sostituzione parziale della protesi. I test di laboratorio hanno evidenziato una neutropenia persistente, nonostante il ritiro di farmaci mielotossici e il miglioramento della sepsi, raggiungendo 0neutrofili/mm3. Retrospettivamente, quando ha esaminato il numero di neutrofili, ha avuto bassi conteggi un anno prima, tra 1800 e 1000/mm3. Il resto della conta ematica e della biochimica erano normali. L’ESR era 80mm / h e CRP 111mg / l. Ha mantenuto alti livelli di fattore reumatoide (6930U/ml) e anti-CCP (300U/ml) e ha anche avuto ipergammaglobulinemia policlonale. Gli anticorpi antinucleari e gli antigeni nucleari estraibili erano negativi e i livelli di complemento rientravano nei valori normali. HLA tipizzazione ha mostrato che portava l’aplotipo DRB1 * 0404 (DR4) e splenomegalia lieve è stato rilevato (13,7 cm) sulla tomografia computerizzata addominale.

Pneumoconiosi e noduli reumatoidi polmonari.
Lo striscio di sangue periferico mostrava linfocitosi da LGL (Fig. 2), che nell’immunofenotipo corrispondeva al 42% dei leucociti totali, con un fenotipo aberrante di linfociti T citotossici (CD3+, CD8+, CD5+, CD7+/−, CD4−, CD56− e DR+). La biopsia del midollo osseo ha mostrato il 20% della cellularità totale del midollo corrispondente alla stessa espansione clonale (confermata dal riarrangiamento della regione variabile della gamma TCR). Sulla base di questi risultati, è stata fatta una diagnosi di LGLL e il trattamento con metotrexato 15 mg alla settimana è stato riavviato nonostante 3 mesi con neutropenia persistente (3
), richiedendo la somministrazione frequente di fattori stimolanti la granulopoiesi. Successivamente è stato trattato con ciclofosfamide, vincristina e prednisone ad alte dosi. Dopo 6 mesi di trattamento, la neutropenia persiste.
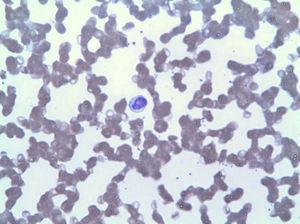
Linfocitosi dovuta a LGL.
Discussione
LGLL è una leucemia cronica caratterizzata dall’espansione del fenotipo monoclonale LGL di linfociti T citotossici attivati o meno frequentemente cellule NK.5 L’età media della diagnosi è di 60 anni ed è frequentemente associata a malattie autoimmuni, in particolare AR, ma è stata descritta anche nella colite ulcerosa, nella sindrome di Sjögren, nel lupus eritematoso e nella sclerosi multipla 1. I pazienti con AR che hanno associato LGLL hanno una presentazione clinica simile a quella di FS. Di solito sono pazienti con AR di lunga data, gravi danni articolari e conseguenze significative e aumento della frequenza di manifestazioni extra-articolari come noduli reumatoidi, linfoadenopatia, ulcere pretibiali, pleurite, pigmentazione cutanea, neuropatia o episclerite.6 Il paziente aveva anche un’altra rara complicanza di AR, pneumoconiosi reumatoide o sindrome di Caplan, caratterizzata dalla comparsa di noduli polmonari con istopatologia simile a quella dei noduli reumatoidi tipici in pazienti con una storia di esposizione professionale a polveri inorganiche come silice, carbone o granito.7 A nostra conoscenza, il presente caso è il primo che descrive la presentazione della sindrome di Caplan e della LGLL nello stesso paziente. Nella maggior parte dei casi, la presentazione LGLL è grave neutropenia associata a infezioni batteriche ricorrenti. I microrganismi più comunemente coinvolti sono Staphylococcus aureus, Streptococcus spp. e bacilli gram-negativi. Meno comunemente anemia, febbre, sudorazione notturna e ingrossamento del fegato e della splena5 possono anche accompagnarlo. Fino a un terzo dei pazienti con LLGG non ha alcuna attività clinica AR apparente al momento della diagnosi, ma mantiene alti livelli di ESR.6 Fino al 40% dei pazienti con FS ha linfocitosi LGL.7 Questo fatto, insieme alla somiglianza clinica e all’associazione con HLA-DR4 ha fortemente suggerito che FS e LGLL associati a RA sono espressioni della stessa entità caratterizzata dalla proliferazione di LGL.8 Altre forme includono anche forme più lievi come linfocitosi reattiva e infezioni a forme più aggressive di NK5 LGLL. La diagnosi LGLL si basa sul ritrovamento di un’espansione monoclonale di LGL nel sangue periferico e nel midollo osseo con un immunofenotipo caratteristico (CD3+, CD4−, CD8 +, CD16+, CD28− e CD57+). La clonalità è confermata dallo studio del gene reTCR9. In generale, la LGL ha una progressione cronica e indolente, con una sopravvivenza media di 10 anni.1 In rari casi, specialmente quando l’espansione è dovuta a LGL con fenotipo NK, questa leucemia può comportarsi in modo più aggressivo.5 L’indicazione più comune per il trattamento sono infezioni ricorrenti e, meno frequentemente, anemia, splenomegalia sintomatica o comparsa di gravi sintomi B B1.
Il trattamento di prima linea in LGLL sono farmaci immunosoppressori da soli, in particolare metotrexato (10 mg/settimana), ciclosporina A (1-1, 5 mg/kg/2 volte al giorno) o ciclofosfamide orale (50-100 mg/die). Questo trattamento è efficace in circa il 50% dei pazienti, ottenendo la correzione delle citopenie, ma non sradicando le cellule leucemiche.1 I glucocorticoidi possono essere utilizzati per accelerare la risposta e i fattori stimolanti la granulopoiesi sono utili nella gestione iniziale della neutropenia. In pazienti refrattari e in quelli con presentazione molto aggressiva il trattamento con regimi chemioterapici simili a CHOP (ciclofosfamide, vincristina, doxorubicina e prednisone) e altri schemi per il linfoma sono stati provati, ma non hanno chiaramente dimostrato la loro efficacia. Altri trattamenti che sono stati testati sono analoghi delle purine, Alemtuzumab, bortezomib, splenectomia e trapianto allogenico di midollo osseo, con risultati variabili.3
Conclusioni
Sia FS che LGLL sono complicazioni rare di RA, che compaiono nella malattia di lunga data, con danni strutturali significativi e manifestazioni extra-articolari. Nei pazienti con AR e neutropenia di lunga durata deve essere esclusa la presenza di proliferazioni clonali di LGL nel sangue periferico e/o nel midollo osseo, consentendo la diagnosi di LGLL. Il trattamento di prima linea è l’uso di farmaci immunosoppressori, come il metotrexato a basse dosi, e i glucocorticoidi possono essere associati a fattori stimolanti la granulopoiesi. Altre modalità di trattamento come la chemioterapia o la splenectomia hanno mostrato risultati variabili in alcuni casi refrattari.
Informazioni etiche
Protezione di soggetti umani e animali. Gli autori dichiarano che non sono stati eseguiti esperimenti su esseri umani o animali per questa indagine.
Riservatezza dei dati. Gli autori dichiarano di aver seguito i protocolli del loro centro di lavoro sulla pubblicazione dei dati dei pazienti e che tutti i pazienti inclusi nello studio hanno ricevuto informazioni sufficienti e hanno dato il loro consenso informato per iscritto a partecipare a tale studio.