idiopatisk Livedo Racemosa med splenomegali och diffus lymfadenopati
Sneddon syndrom (SS) beskrevs först 1965 hos patienter med ihållande livedo racemosa och neurologiska händelser.1 eftersom de andra manifestationerna av SS är ospecifika (t.ex. hypertoni, hjärtvalvulopati, arteriell och venös ocklusion) försenas diagnosen ofta. Många patienter som upplever prodromala neurologiska symtom som huvudvärk, depression, ångest, yrsel och neuropati förekommer ofta hos en läkare innan de utvecklar ischemiska hjärnmanifestationer2 men får sällan rätt diagnos. Uppkomsten av cerebrala ocklusiva händelser uppträder vanligtvis hos patienter yngre än 45 år och kan förekomma som en övergående ischemisk attack, stroke eller intrakraniell blödning.3 sjukdomen är vanligare hos kvinnor än män (2:1-förhållande). Den exakta patogenesen av SS är fortfarande okänd, och även om det har betraktats som en separat enhet från systemisk lupus erythematosus och andra antifosfolipidstörningar, har det antagits att en immunologisk dysfunktion skadar kärlväggar som leder till trombos.
kutana fynd associerade med SS involverar små till medelstora dermal-subdermala artärer. Histopatologi hos vissa patienter visar proliferation av endotel-och fibrinavlagringar med efterföljande utplåning av involverade artärer.4 hos många patienter inklusive vår patient misslyckas histopatologisk undersökning av involverad hud att visa specifika avvikelser.1 Zelger et al5 rapporterade sekvensen av histopatologiska hudhändelser i en serie antifosfolipidnegativa SS-patienter. Författarna rapporterade att endast små artärer vid dermis-subcutis-korsningen var inblandade och en progression av endoteldysfunktion observerades. Författarna trodde att det fanns flera icke-specifika steg före fibrinocklusion av involverade artärer.5 steg i involverade lossning av endotelceller med ospecifik perivaskulär lymfocytisk infiltration med perivaskulär inflammation och lymfocytisk infiltration som representerar sjukdomens främsta drivkraft.5,6 detta stadium anses vara kortlivat, varför det har blivit oupptäckt i många år hos SS-patienter. Steg II till IV utvecklas genom fibrinavsättning och ocklusion.5 histologiska egenskaper i steg i till II har inte rapporterats på grund av sen diagnos av SS. Steg i-patienter uppvisar vanligtvis en genomsnittlig varaktighet av symtom på 6 månader med få neurologiska symtom, den vanligaste är parestesi i benen.5
fallrapport
en 37-årig kvinna med epigastrisk ömhet på vänster sida och splenomegali sett på datortomografi hänvisades av en hematolog för utvärdering av ett retikulärt utslag på vänster sida av flanken av 9 månaders varaktighet med en antagen diagnos av fokal melanoderma. Hennes medicinska historia var anmärkningsvärd för en medfödd ventrikulär septal defekt och koarktation av aorta, liksom endometrios, myalgi och ledstyvhet som alla hade utvecklats under det senaste året. Hennes sjukdomshistoria var också anmärkningsvärt för njursten, colon irritabile, och kronisk bihåleinflammation, samt psykiatrisk depression och ångest. Hon hade nyligen diagnostiserats med måttlig hypertoni och hade haft svårt att bli gravid under de senaste åren med 3 på varandra följande missfall under första trimestern. Neurologiska symtom inkluderade neuropati som involverade fötterna, intermittent parestesi i benen och en historia av kronisk migränhuvudvärk i flera månader.
dermatologisk undersökning avslöjade en något överviktig kvinna med ett 25-20-cm-skumma, erytematösa, oregelbundna, nätliknande mönster på vänster sida av övre och nedre stammen (Figur 1). Omfattande livedo racemosa förändrades inte av temperaturförändringar och hade varit oförändrad i mer än 9 månader. Det fanns inga tecken på klåda eller sår, och områden med livedo racemosa var något ömma för palpation.
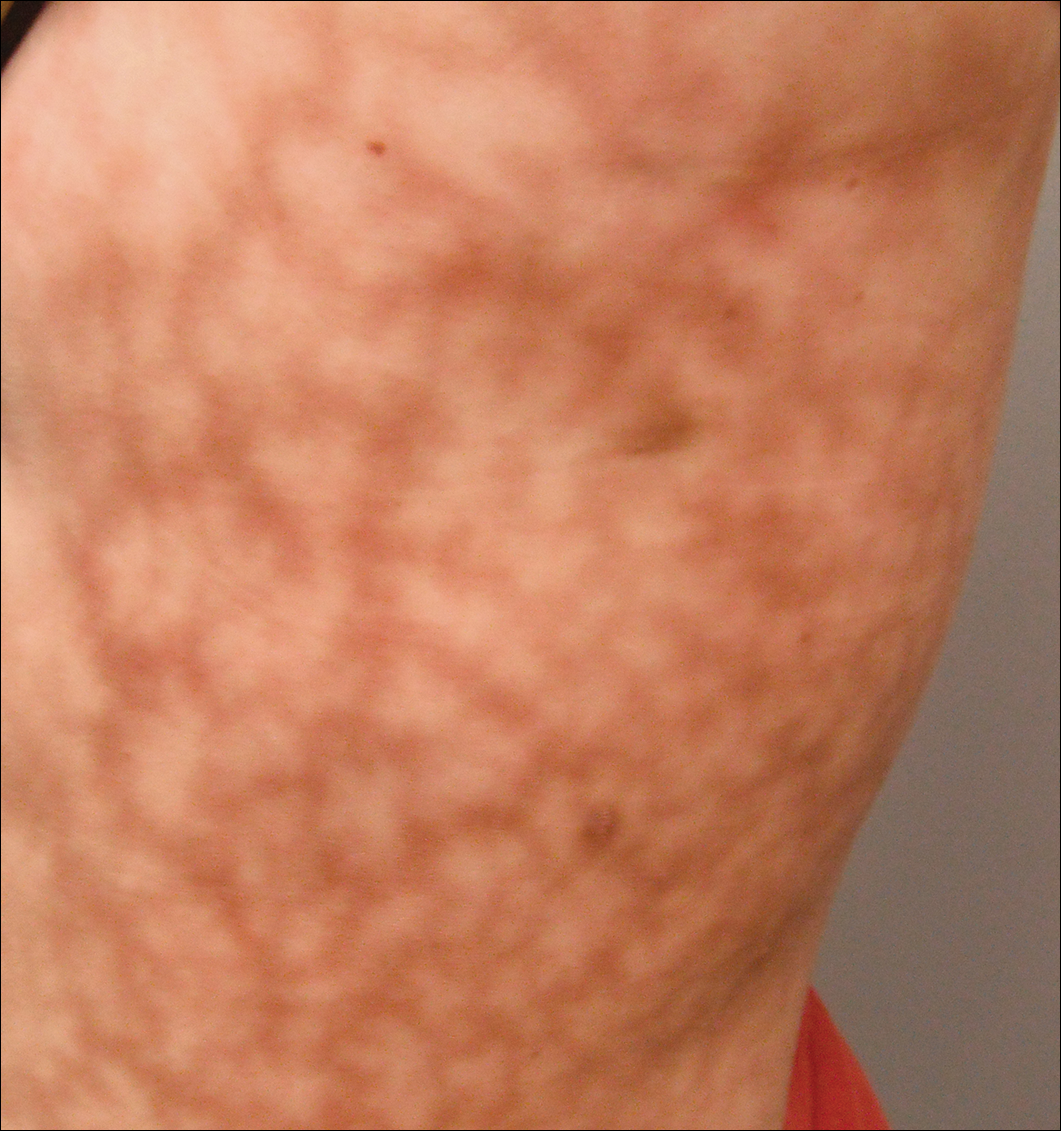
Figur 1. Livedo racemosa presentera som en netlike violaceous mönster på den vänstra sidan av stammen mäter 25 20 xnumx xnumx cm.
Vi utförde 2 uppsättningar av tre 4 mm biopsier. Den första uppsättningen riktade områden inom violaceous mönstret, medan den andra uppsättningen riktade områden med normal vävnad mellan de fläckiga områdena. Alla 6 prover visade ytlig perivaskulär lymfocytisk infiltrera utan tecken på vaskulit eller bindvävssjukdom. Fartygen visade ingen mikrotrombi eller omgivande fibros. Inga eosinofiler identifierades i epidermis. Det fanns inga tecken på ökat dermal mucin. Både de ytliga och djupa vaskulära plexuserna var obemärkliga och visade inga tecken på skador på väggarna (Figur 2).
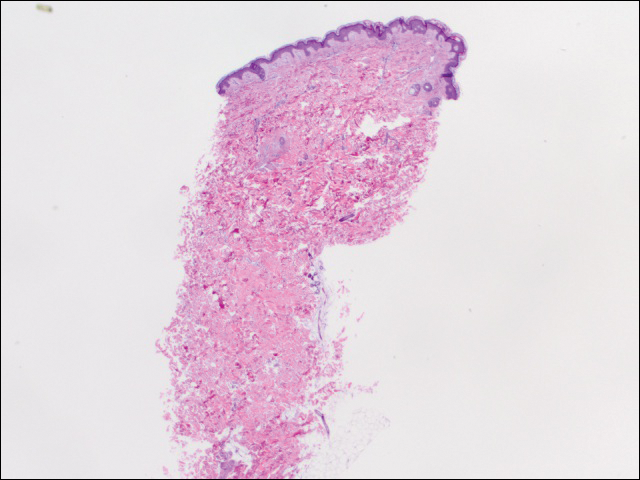
Figur 2. Punchbiopsi från vänster sida av stammen visade fokal melanoderma och gles ytlig perivaskulär lymfocytisk infiltrering utan tecken på vaskulit, mikrotrombi eller fibrinavsättning (H&E, original förstoring 20 i 20).
för att utesluta andra möjliga orsaker till livedo racemosa utfördes fullständigt blodcellsantal, omfattande metabolisk panel, koagulationsprofil, lipastest, urinanalys, serologisk testning och immunologisk upparbetning. Lipas var inom referensområdet. Det fullständiga antalet blodkroppar avslöjade mild anemi, medan resten av värdena låg inom referensområdet. En immunologisk upparbetning inkluderade Sjabbiggrens syndromantigen a, Sjabbiggrens syndromantigen B, antikardiolipinantikroppar och antinukleär antikropp, som alla var negativa. Familjehistoria var anmärkningsvärd för första gradens släktingar med systemisk lupus erythematosus och Crohns sjukdom.
datortomografi avslöjade förstoring av mjälten, såväl som periaortisk, portacaval och porta hepatis lymfadenopati. Baserat på laboratoriefynd och klinisk presentation samt patientens medicinska historia var diagnosen uteslutning idiopatisk livedo racemosa med okänd progression till fullblåst SS. Patienten uppfyllde inte de nuvarande diagnostiska kriterierna för SS, och hennes immunologiska studier misslyckades med att bekräfta några nuvarande antikroppar, men involvering av retikuloendotelialsystemet pekade på produktion av antikroppar som ännu inte kunde detekteras vid laboratorietestning.