Racemosa Livedo Idiopática Con Esplenomegalia y Linfadenopatía Difusa
El síndrome de Sneddon (SS) se describió por primera vez en 1965 en pacientes con racemosa livedo persistente y acontecimientos neurológicos.1 Debido a que las otras manifestaciones de SS son inespecíficas (por ejemplo, hipertensión, valvulopatía cardíaca, oclusión arterial y venosa), el diagnóstico a menudo se retrasa. Muchos pacientes que experimentan síntomas neurológicos prodrómicos, como dolores de cabeza, depresión, ansiedad, mareos y neuropatía, a menudo se presentan a un médico antes de desarrollar manifestaciones cerebrales isquémicas2, pero rara vez reciben el diagnóstico correcto. La aparición de eventos oclusivos cerebrales ocurre típicamente en pacientes menores de 45 años y puede presentarse como un ataque isquémico transitorio, accidente cerebrovascular o hemorragia intracraneal.3 La enfermedad es más prevalente en mujeres que en hombres (proporción 2:1). La patogénesis exacta del SS es aún desconocida, y aunque se ha considerado como una entidad separada del lupus eritematoso sistémico y otros trastornos antifosfolípidos, se ha postulado que una disfunción inmunológica daña las paredes de los vasos que conducen a la trombosis.
Los hallazgos cutáneos asociados con SS involucran arterias dérmicas subdérmicas de tamaño pequeño a mediano. La histopatología en algunos pacientes demuestra proliferación de depósitos de endotelio y fibrina con posterior obliteración de las arterias comprometidas.4 En muchos pacientes, incluido el nuestro, el examen histopatológico de la piel afectada no muestra anomalías específicas.1 Zelger et al5 relataron la secuencia de eventos histopatológicos de la piel en una serie de pacientes con SS antifosfolípidos negativos. Los autores relataron que solo las arterias pequeñas en la unión dermis-subcutis estaban comprometidas y se observó una progresión de la disfunción endotelial. Los autores creían que había varios estadios inespecíficos antes de la oclusión de fibrina de las arterias comprometidas.El estadio 5 I involucró el aflojamiento de las células endoteliales con infiltración linfocítica perivascular inespecífica con inflamación perivascular e infiltración linfocítica que representan el motor principal de la enfermedad.5,6 Se cree que esta etapa es de corta duración, por lo que ha pasado desapercibida durante muchos años en pacientes con SS. Los estadios II a IV progresan a través de la deposición y oclusión de fibrina.5 No se notificaron las características histológicas de los estadios I a II debido al diagnóstico tardío de SS. Los pacientes en estadio I generalmente presentan una duración promedio de los síntomas de 6 meses con pocos síntomas neurológicos, el más común es la parestesia de las piernas.5
Reporte de un caso
Una mujer de 37 años con sensibilidad epigástrica en el lado izquierdo y esplenomegalia observada en tomografía computarizada fue remitida por un hematólogo para la evaluación de una erupción reticular en el lado izquierdo del costado de 9 meses de duración con un diagnóstico presunto de melanoderma focal. Su historial médico fue notable por una comunicación interventricular congénita y coartación de la aorta, así como endometriosis, mialgia y rigidez articular que se habían desarrollado durante el último año. Su historial médico también fue notable por nefrolitiasis, síndrome del intestino irritable y sinusitis crónica, así como depresión psiquiátrica y trastornos de ansiedad. Recientemente le habían diagnosticado hipertensión moderada y había experimentado dificultades para quedar embarazada durante los últimos años con 3 abortos espontáneos consecutivos en el primer trimestre. Los síntomas neurológicos incluyeron neuropatía en los pies, parestesia intermitente de las piernas y antecedentes de migrañas crónicas durante varios meses.
El examen dermatológico reveló una mujer con sobrepeso leve, con un patrón de 25×30 cm de color oscuro, eritematoso, irregular, en forma de red en el lado izquierdo de la parte superior e inferior del tronco (Figura 1). El livedo racemosa extenso no se alteró por los cambios de temperatura y había permanecido inalterado durante más de 9 meses. No hubo signos de prurito o ulceraciones, y las zonas de livedo racemosa estaban ligeramente sensibles a la palpación.
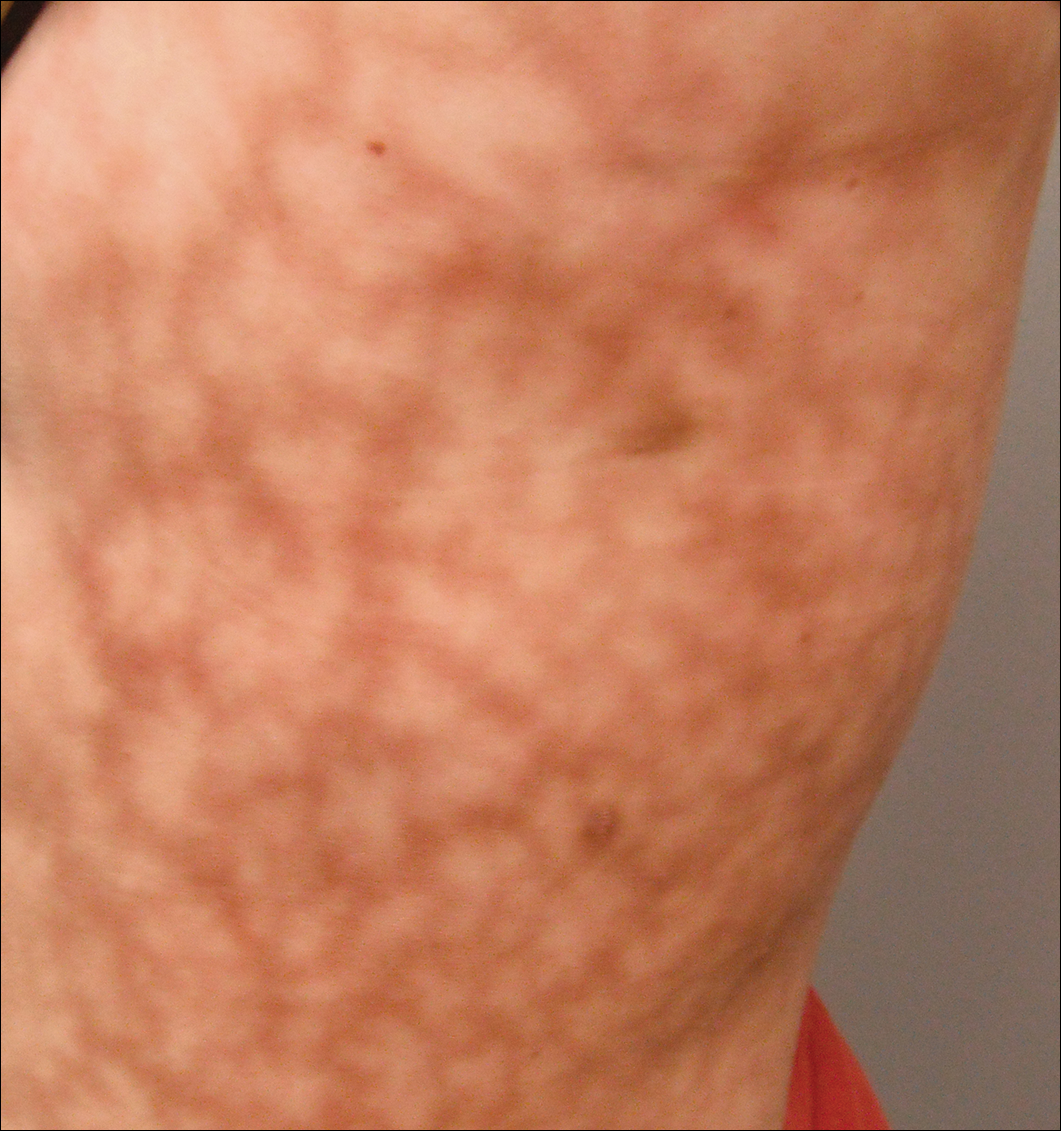
la Figura 1. Livedo racemosa presenta un patrón violáceo en forma de red en el lado izquierdo del tronco de 25×30 cm.
se realizaron 2 grupos de tres, de 4 mm de biopsias. El primer conjunto apuntó a áreas dentro del patrón violáceo, mientras que el segundo conjunto apuntó a áreas de tejido normal entre las áreas moteadas. Las 6 muestras mostraron infiltrado linfocítico perivascular superficial sin evidencia de vasculitis o enfermedad del tejido conectivo. Los vasos no mostró microtrombos o alrededor de la fibrosis. No se identificaron eosinófilos dentro de la epidermis. No hubo evidencia de aumento de mucina dérmica. Tanto los plexos vasculares superficiales como los profundos eran poco notables y no mostraban evidencia de daño en las paredes (Figura 2).
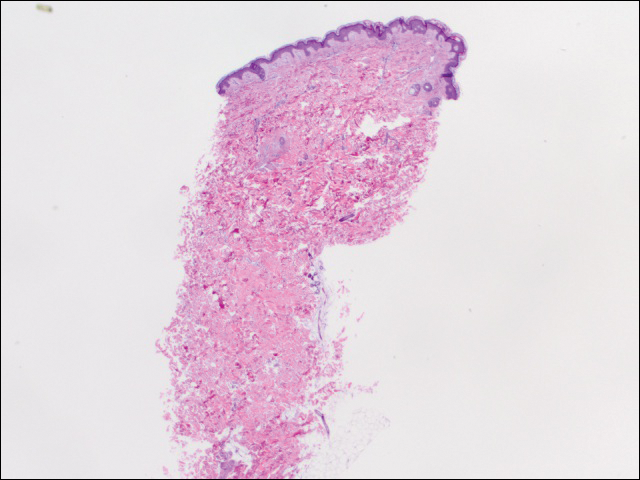
la Figura 2. La biopsia con sacabocados del lado izquierdo del tronco mostró melanoderma focal e infiltrado linfocítico perivascular superficial escaso sin evidencia de vasculitis, microtrombis o deposición de fibrina (H&E, aumento original ×20).
Para descartar otras posibles causas de livedo racemosa, hemograma completo, grupo de pruebas metabólicas completas, perfil de coagulación, de la lipasa de prueba, análisis de orina, pruebas serológicas, y inmunológica diagnóstico diferencial se realiza. La lipasa estaba dentro del rango de referencia. El recuento completo de células sanguíneas reveló anemia leve, mientras que el resto de los valores estaban dentro del rango de referencia. Un examen inmunológico incluyó antígeno A del síndrome de Sjögren, antígeno B del síndrome de Sjögren, anticuerpos anticardiolipina y anticuerpos antinucleares, todos negativos. Los antecedentes familiares fueron notables para parientes de primer grado con lupus eritematoso sistémico y enfermedad de Crohn.
La tomografía computarizada reveló agrandamiento del bazo, así como linfadenopatía periaórtica, portacaval y porta hepatis. Con base en los hallazgos de laboratorio y la presentación clínica, así como en la historia clínica del paciente, el diagnóstico de exclusión fue livedo racemosa idiopático con progresión desconocida a SS completa. La paciente no cumplía los criterios diagnósticos actuales de SS, y sus estudios inmunológicos no confirmaron la presencia de anticuerpos, pero la afectación del sistema reticuloendotelial apuntaba a la producción de anticuerpos que aún no eran detectables en las pruebas de laboratorio.