La Leucemia de Linfocitos Granulares Grandes como Complicación de la Artritis Reumatoide | Reumatología Clínica
Introducción
El síndrome de Felty (FS) es una complicación sistémica poco frecuente (menos del 1%) de la artritis reumatoide (AR), caracterizada por la tríada de la AR, la neutropenia persistente (3
) y la esplenomegalia de tamaño variable que puede variar desde esplenomegalia subclínica, solo detectable por imágenes, hasta esplenomegalia masiva1. Se presenta principalmente en casos de larga data con enfermedad articular grave y manifestaciones extraarticulares, y tiene una fuerte asociación con el haplotipo HLA-DR4 (casi el 95% de los casos).2 En 30-40% de los pacientes con FS hay una expansión de linfocitos granulares grandes (LGL).1 Las LGL representan el 10-15% de las células mononucleares circulantes y se identifican morfológicamente por su gran tamaño (15-18 µm), su núcleo redondo o indentado y abundante citoplasma con gránulos azurofílicos. El fenotipo de estas células puede ser linfocito T citotóxico (CD8+, CD57+) o asesino natural (NK) (CD3−, CD8−, CD56+).3 Cuando la expansión de la LGLG es monoclonal y se asocia con infiltración de la médula ósea y el bazo por parte de estas células, se denomina leucemia linfocítica granular grande (LGLL) y se considera un trastorno linfoproliferativo crónico de bajo grado. Su presentación clínica es similar a la del FS, destacando la mayor susceptibilidad a infecciones bacterianas asociadas a neutropenia, anemia y esplenomegalia, como también se ha denominado «pseudo-Felty».3,4 Presentación clínica
Un trabajador jubilado de 70 años de una cantera de granito fue diagnosticado a los 43 años de edad con AR seropositiva con afectación de manos, pies, rodillas y caderas. Posteriormente desarrolló neumoconiosis y nódulos reumatoides pulmonares (Fig. 1), siendo diagnosticado como síndrome de Caplan. Durante su progresión, fue tratado con AINE, corticosteroides, sales de oro, ciclosporina y metotrexato. A pesar de ello, el paciente desarrolló daño estructural en manos, pies y caderas, requiriendo la colocación de prótesis en ambas caderas a los 51 y 54 años de edad, respectivamente. En los últimos años, su enfermedad se mantuvo estable, tratada con metotrexato 10 mg por semana y dosis bajas de glucocorticoides, sin evidencia de actividad inflamatoria articular. Presentaba deformidades de cuello de cisne en todos los dedos y nódulos reumatoides en los codos. hace 12 meses, de repente, desarrolló fiebre y dolor en la ingle derecha, con un shock séptico posterior, lo que demuestra una infección de la cadera derecha por Salmonella spp. Se suspendieron metotrexato y glucocorticoides, y se le trató con terapia antibiótica prolongada y reemplazo parcial de la prótesis. Las pruebas de laboratorio evidenciaron neutropenia persistente, a pesar de la retirada de fármacos mielotóxicos y la mejoría de la sepsis, alcanzando 0neutrofilos/mm3. Retrospectivamente, al revisar el número de neutrófilos, tenía recuentos bajos un año antes, entre 1800 y 1000/mm3. El resto del hemograma y la bioquímica eran normales. La VSG fue de 80 mm / h y PCR de 111 mg/l. Mantuvo altos niveles de factor reumatoide (6930U/ml) y anti-CCP (300U / ml) y también tuvo hipergammaglobulinemia policlonal. Los anticuerpos antinucleares y antígenos nucleares extraíbles fueron negativos y los niveles de complemento estuvieron dentro de los valores normales. La tipificación de HLA mostró que portaba el haplotipo DRB1*0404 (DR4) y se detectó esplenomegalia leve (13,7 cm) en tomografía computarizada abdominal.

Neumoconiosis y nódulos reumatoides pulmonares.
El frotis de sangre periférica mostró linfocitosis por LGL (Fig. 2), que en el inmunofenotipo correspondió al 42% del total de leucocitos, con un fenotipo aberrante de linfocitos T citotóxicos (CD3+, CD8+, CD5+, CD7+/−, CD4−, CD56− y DR+). La biopsia de médula ósea mostró un 20% de la celularidad medular total correspondiente a la misma expansión clonal (confirmada por el reordenamiento de la región variable de la gamma TCR). En base a estos hallazgos, se realizó un diagnóstico de LGLL y se reinició el tratamiento con metotrexato 15 mg semanal a pesar de 3 meses con neutropenia persistente (3
), que requirió la administración frecuente de factores estimulantes de granulopoyesis. Posteriormente fue tratado con ciclofosfamida, vincristina y prednisona en dosis altas. Después de 6 meses de tratamiento, la neutropenia persiste.
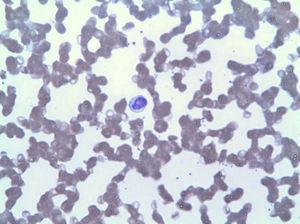
Linfocitosis debida a LGL.
Discusión
La LGLL es una leucemia crónica caracterizada por la expansión del fenotipo monoclonal LGLL de linfocitos T citotóxicos activados o, con menos frecuencia, de células NK.5 La edad promedio de diagnóstico es de 60 años y se asocia con frecuencia a enfermedades autoinmunes, en particular AR, pero también se ha descrito en colitis ulcerosa, síndrome de Sjögren, lupus eritematoso y esclerosis multiple1. Los pacientes con AR que tienen LGLL asociada tienen una presentación clínica similar a la del SM. Por lo general, son pacientes con artritis reumatoide de larga duración, daño articular grave y consecuencias significativas, y aumento de la frecuencia de manifestaciones extraarticulares como nódulos reumatoides, linfadenopatía, úlceras pretibiales, pleuritis, pigmentación de la piel, neuropatía o episcleritis.6 El paciente también tenía otra complicación rara de AR, neumoconiosis reumatoide o síndrome de Caplan, caracterizada por la aparición de nódulos pulmonares con histopatología similar a la de los nódulos reumatoides típicos en pacientes con antecedentes de exposición ocupacional a polvos inorgánicos como sílice, carbón o granito.7 Hasta donde sabemos, el presente caso es el primero que describe la presentación del síndrome de Caplan y LGLL en el mismo paciente. En la mayoría de los casos, la presentación de LGLL es neutropenia grave asociada con infecciones bacterianas recurrentes. Los microorganismos más comúnmente involucrados son Staphylococcus aureus, Streptococcus spp. y bacilos gramnegativos. Con menos frecuencia, la anemia, la fiebre, los sudores nocturnos y el agrandamiento del hígado y el bazo también pueden acompañarla.5 Hasta un tercio de los pacientes con GGL no tienen actividad clínica aparente de artritis reumatoide en el momento del diagnóstico, pero mantienen niveles altos de VSG.6 Hasta el 40% de los pacientes con FS tienen linfocitosis de LGL.7 Este hecho, junto con la similitud clínica y la asociación con HLA-DR4, ha sugerido fuertemente que el FS y la LGLL asociadas a AR son expresiones de la misma entidad caracterizada por la proliferación de LGLL.8 Otras formas incluyen también formas más leves, como linfocitosis reactiva e infecciones a formas más agresivas de LGLL NK5. El diagnóstico de LGLL se basa en el hallazgo de una expansión monoclonal de LGLL en sangre periférica y médula ósea con un inmunofenotipo característico (CD3+, CD4−, CD8 +, CD16+, CD28− y CD57+). La clonalidad se confirma estudiando el gen reTCR9. En general, la LGLI tiene una progresión crónica e indolente, con una supervivencia media de 10 años.1 En casos raros, especialmente cuando la expansión se debe a LGL con fenotipo NK, esta leucemia puede comportarse de manera más agresiva.5 Las indicaciones de tratamiento más frecuentes son las infecciones recurrentes y, con menor frecuencia, la anemia, la esplenomegalia sintomática o la aparición de síntomas B graves B1.
El tratamiento de primera línea en LGLL son medicamentos inmunosupresores solos, específicamente metotrexato (10 mg/semana), ciclosporina A (1-1, 5 mg/kg/2 veces al día) o ciclofosfamida oral (50-100 mg/día). Este tratamiento es efectivo en aproximadamente el 50% de los pacientes, logrando la corrección de las citopenias, pero no erradicando las células leucémicas.1 Los glucocorticoides se pueden usar para acelerar la respuesta y los factores estimulantes de la granulopoyesis son útiles en el manejo inicial de la neutropenia. En pacientes refractarios y en aquellos con presentación muy agresiva, se ha probado el tratamiento con regímenes de quimioterapia similares a CHOP (ciclofosfamida, vincristina, doxorrubicina y prednisona) y otros esquemas para el linfoma, pero no han demostrado claramente su eficacia. Otros tratamientos que se han probado son análogos de purina, Alemtuzumab, bortezomib, esplenectomía y trasplante alogénico de médula ósea, con resultados variables.3
Conclusiones
Tanto el FS como la LGLL son complicaciones raras de la AR, que aparecen en la enfermedad de larga data, con daño estructural significativo y manifestaciones extraarticulares. En pacientes con artritis reumatoide de larga duración y neutropenia, se debe descartar la presencia de proliferaciones clonales de LGLI en sangre periférica y/o médula ósea, lo que permite el diagnóstico de LGLI. El tratamiento de primera línea es el uso de medicamentos inmunosupresores, como metotrexato en dosis bajas, y los glucocorticoides pueden estar asociados con factores estimulantes de la granulopoyesis. Otras modalidades de tratamiento, como la quimioterapia o la esplenectomía, han mostrado resultados variables en algunos casos refractarios.
Divulgación ética
Protección de sujetos humanos y animales. Los autores declaran que no se realizaron experimentos en seres humanos o animales para esta investigación.
Confidencialidad de los Datos. Los autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes y que todos los pacientes incluidos en el estudio han recibido información suficiente y han dado su consentimiento informado por escrito para participar en dicho estudio.