Livedo Racemosa Idiopathique Présentant une Splénomégalie et une Lymphadénopathie diffuse
Le syndrome de Sneddon (SS) a été décrit pour la première fois en 1965 chez des patients présentant un livedo racemosa persistant et des événements neurologiques.1 Parce que les autres manifestations de SS ne sont pas spécifiques (par exemple, hypertension, valvulopathie cardiaque, occlusion artérielle et veineuse), le diagnostic est souvent retardé. De nombreux patients présentant des symptômes neurologiques prodromiques tels que maux de tête, dépression, anxiété, vertiges et neuropathie se présentent souvent à un médecin avant de développer des manifestations cérébrales ischémiques 2, mais reçoivent rarement le diagnostic correct. L’apparition d’événements occlusifs cérébraux survient généralement chez les patients de moins de 45 ans et peut se présenter sous la forme d’un accident ischémique transitoire, d’un accident vasculaire cérébral ou d’une hémorragie intracrânienne.3 La maladie est plus répandue chez les femmes que chez les hommes (rapport 2:1). La pathogenèse exacte de la SS est encore inconnue, et bien qu’elle ait été considérée comme une entité distincte du lupus érythémateux disséminé et d’autres troubles antiphospholipidiques, il a été postulé qu’un dysfonctionnement immunologique endommage les parois des vaisseaux conduisant à une thrombose.
Les résultats cutanés associés à la SS concernent des artères dermo-sous-dermiques de petite à moyenne taille. L’histopathologie chez certains patients montre une prolifération des dépôts d’endothélium et de fibrine avec oblitération ultérieure des artères impliquées.4 Chez de nombreux patients, y compris notre patient, l’examen histopathologique de la peau impliquée ne montre pas d’anomalies spécifiques.1 Zelger et al5 ont rapporté la séquence d’événements cutanés histopathologiques chez une série de patients SS antiphospholipides négatifs. Les auteurs ont rapporté que seules de petites artères à la jonction derme-sous-cutis étaient impliquées et une progression du dysfonctionnement endothélial a été observée. Les auteurs pensaient qu’il y avait plusieurs stades non spécifiques avant l’occlusion de la fibrine des artères impliquées.Le stade 5 I impliquait un relâchement des cellules endothéliales avec infiltration lymphocytaire périvasculaire non spécifique avec inflammation périvasculaire et infiltration lymphocytaire représentant le moteur principal de la maladie.5,6 On pense que ce stade est de courte durée, ce qui explique pourquoi il est passé inaperçu pendant de nombreuses années chez les patients SS. Les stades II à IV progressent par dépôt de fibrine et occlusion.5 Les caractéristiques histologiques des stades I à II n’ont pas été rapportées en raison d’un diagnostic tardif de SS. Les patients de stade I présentent généralement une durée moyenne de symptômes de 6 mois avec peu de symptômes neurologiques, le plus fréquent étant la paresthésie des jambes.5
Rapport de cas
Une femme de 37 ans présentant une sensibilité épigastrique du côté gauche et une splénomégalie observée par tomodensitométrie a été référée par un hématologue pour une évaluation d’une éruption réticulaire du côté gauche du flanc d’une durée de 9 mois avec un diagnostic présumé de mélanodermie focale. Ses antécédents médicaux étaient remarquables pour une anomalie septale ventriculaire congénitale et une coarctation de l’aorte, ainsi qu’une endométriose, une myalgie et une raideur articulaire qui s’étaient toutes développées au cours de la dernière année. Ses antécédents médicaux étaient également remarquables pour la néphrolithiase, le syndrome du côlon irritable et la sinusite chronique, ainsi que la dépression psychiatrique et les troubles anxieux. Elle avait récemment reçu un diagnostic d’hypertension modérée et avait eu des difficultés à tomber enceinte au cours des dernières années avec 3 fausses couches consécutives au premier trimestre. Les symptômes neurologiques comprenaient une neuropathie impliquant les pieds, une paresthésie intermittente des jambes et des antécédents de migraines chroniques pendant plusieurs mois.
L’examen dermatologique a révélé une femme légèrement en surpoids avec un motif sombre, érythémateux, irrégulier et en forme de filet de 25×30 cm sur le côté gauche du tronc supérieur et inférieur (figure 1). Le livedo racemosa étendu n’a pas été modifié par les changements de température et était inchangé depuis plus de 9 mois. Il n’y avait aucun signe de prurit ou d’ulcérations, et les zones de livedo racemosa étaient légèrement sensibles à la palpation.
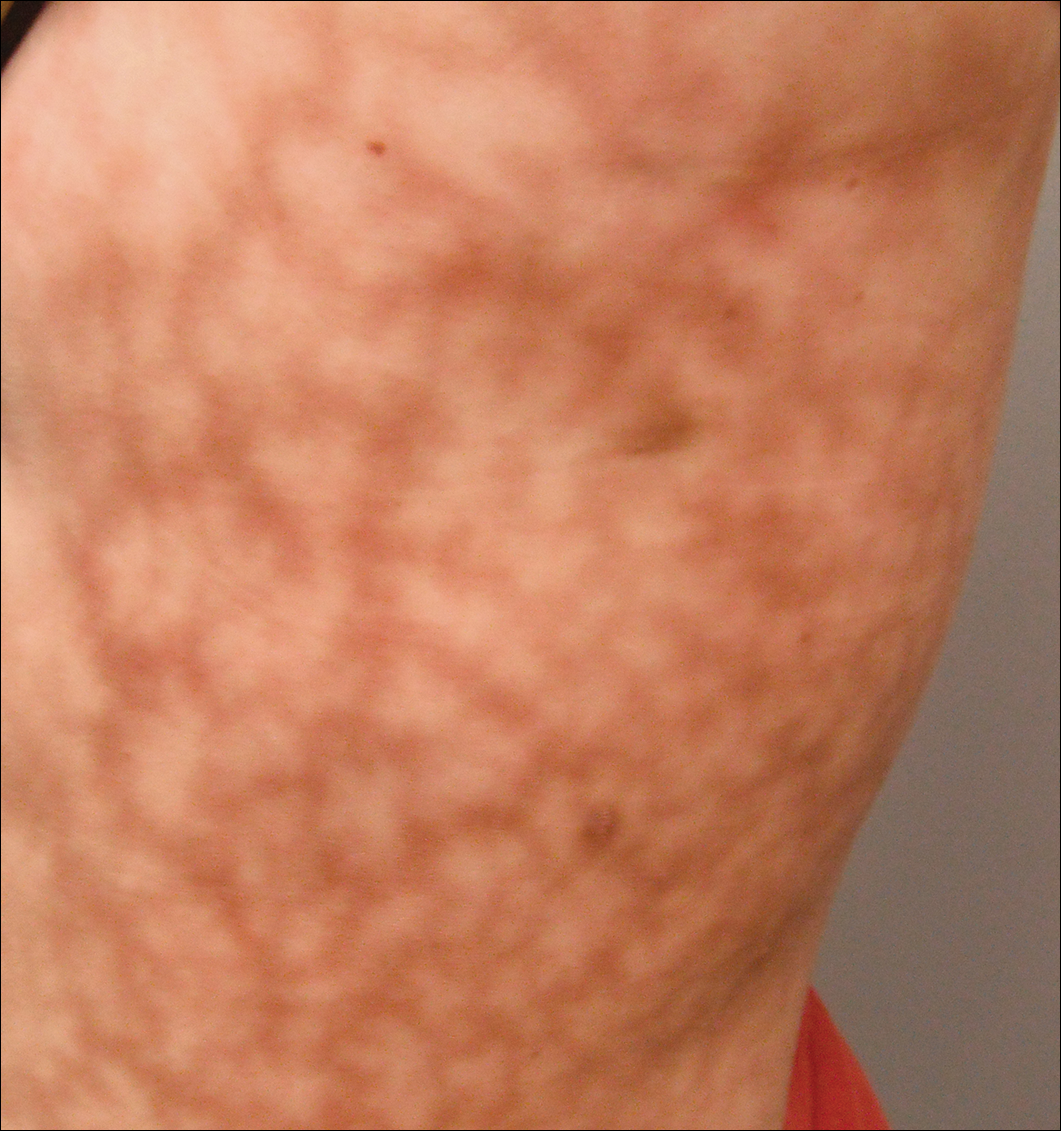
Figure 1. Livedo racemosa se présentant comme un motif violacé en forme de filet sur le côté gauche du tronc mesurant 25×30 cm.
Nous avons effectué 2 séries de trois biopsies de 4 mm. Le premier ensemble a ciblé des zones dans le motif violacé, tandis que le second ensemble a ciblé des zones de tissu normal entre les zones tachetées. Les 6 échantillons présentaient tous un infiltrat lymphocytaire périvasculaire superficiel sans signe de vascularite ou de maladie du tissu conjonctif. Les vaisseaux ne présentaient pas de microthrombi ou de fibrose environnante. Aucun éosinophile n’a été identifié dans l’épiderme. Il n’y avait aucune preuve d’augmentation de la mucine cutanée. Les plexus vasculaires superficiels et profonds n’étaient pas remarquables et ne présentaient aucune preuve de dommages aux parois (figure 2).
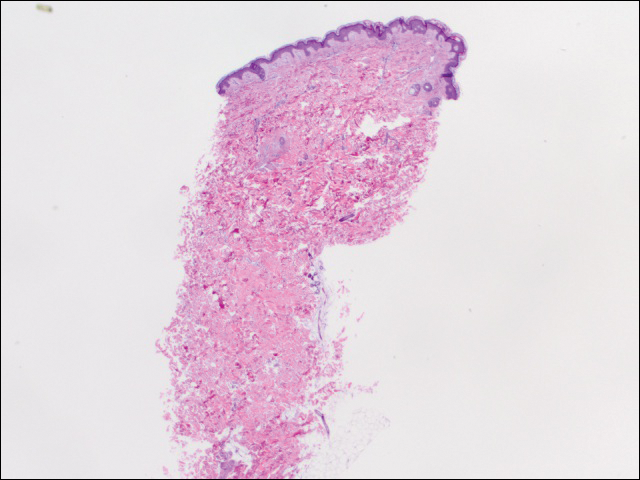
Figure 2. Une biopsie par poinçonnage du côté gauche du tronc a montré une mélanodermie focale et un infiltrat lymphocytaire périvasculaire superficiel clairsemé sans signe de vascularite, de microthrombie ou de dépôt de fibrine (H& E, grossissement original ×20).
Pour exclure d’autres causes possibles de livedo racemosa, une numération globulaire complète, un panel métabolique complet, un profil de coagulation, un test de lipase, une analyse d’urine, des tests sérologiques et un bilan immunologique ont été effectués. La lipase était dans la plage de référence. Le nombre complet de cellules sanguines a révélé une anémie légère, tandis que le reste des valeurs se situait dans la plage de référence. Un bilan immunologique comprenait l’antigène A du syndrome de Sjögren, l’antigène B du syndrome de Sjögren, les anticorps anti-cardiolipines et les anticorps antinucléaires, qui étaient tous négatifs. Les antécédents familiaux étaient remarquables chez les parents au premier degré atteints de lupus érythémateux disséminé et de la maladie de Crohn.
La tomodensitométrie a révélé une hypertrophie de la rate, ainsi qu’une adénopathie périaortique, portacavale et porta hepatis. Sur la base des résultats de laboratoire et de la présentation clinique ainsi que des antécédents médicaux du patient, le diagnostic d’exclusion était livedo racemosa idiopathique avec une progression inconnue vers une SS à part entière. La patiente ne répondait pas aux critères diagnostiques actuels pour la SS, et ses études immunologiques n’ont pas confirmé la présence d’anticorps, mais l’implication du système réticulo-endothélial a mis en évidence la production d’anticorps qui n’étaient pas encore détectables lors des tests de laboratoire.