Livedo Racemosa idiopatica che si presenta con Splenomegalia e linfoadenopatia diffusa
La sindrome di Sneddon (SS) è stata descritta per la prima volta nel 1965 in pazienti con livedo racemosa persistente ed eventi neurologici.1 Poiché le altre manifestazioni di SS sono aspecifiche (ad esempio, ipertensione, valvulopatia cardiaca, occlusione arteriosa e venosa), la diagnosi spesso viene ritardata. Molti pazienti che sperimentano sintomi neurologici prodromici come mal di testa, depressione, ansia, vertigini e neuropatia spesso presentano a un medico prima di sviluppare manifestazioni cerebrali ischemiche2 ma raramente ricevono la diagnosi corretta. L’insorgenza di eventi occlusivi cerebrali si verifica in genere in pazienti di età inferiore ai 45 anni e può presentarsi come attacco ischemico transitorio, ictus o emorragia intracranica.3 La malattia è più diffusa nelle donne rispetto ai maschi (rapporto 2:1). La patogenesi esatta di SS è ancora sconosciuta e sebbene sia stata pensata come un’entità separata dal lupus eritematoso sistemico e da altri disturbi antifosfolipidi, è stato postulato che una disfunzione immunologica danneggia le pareti dei vasi portando alla trombosi.
I risultati cutanei associati a SS coinvolgono arterie dermico – subdermiche di piccole e medie dimensioni. L’istopatologia in alcuni pazienti dimostra la proliferazione dell’endotelio e dei depositi di fibrina con successiva obliterazione delle arterie coinvolte.4 In molti pazienti, incluso il nostro paziente, l’esame istopatologico della pelle coinvolta non mostra anomalie specifiche.1 Zelger et al5 hanno riportato la sequenza di eventi istopatologici cutanei in una serie di pazienti SS antifosfolipidi-negativi. Gli autori hanno riferito che sono state coinvolte solo piccole arterie alla giunzione derma-subcutis ed è stata osservata una progressione della disfunzione endoteliale. Gli autori credevano che ci fossero diversi stadi non specifici prima dell’occlusione della fibrina delle arterie coinvolte.5 Stadio I coinvolto allentamento delle cellule endoteliali con infiltrazione linfocitica perivascolare aspecifica con infiammazione perivascolare e infiltrazione linfocitaria che rappresenta il motore principale della malattia.5,6 Questo stadio è pensato per essere di breve durata, quindi il motivo per cui è passato inosservato per molti anni nei pazienti SS. Gli stadi da II a IV progrediscono attraverso la deposizione e l’occlusione di fibrina.5 Caratteristiche istologiche degli stadi da I a II non sono state riportate a causa della diagnosi tardiva di SS. I pazienti della fase I presentano tipicamente con una durata media dei sintomi di 6 mesi con pochi sintomi neurologici, il più comune è parestesia delle gambe.5
Case Report
Una donna di 37 anni con dolorabilità epigastrica sul lato sinistro e splenomegalia osservata con tomografia computerizzata è stata indirizzata da un ematologo per la valutazione di un’eruzione reticolare sul lato sinistro del fianco della durata di 9 mesi con una presunta diagnosi di melanoderma focale. La sua storia medica era notevole per un difetto congenito del setto ventricolare e coartazione dell’aorta, così come endometriosi, mialgia e rigidità articolare che si erano sviluppate nell’ultimo anno. La sua storia medica era anche notevole per la nefrolitiasi, la sindrome dell’intestino irritabile e la sinusite cronica, così come la depressione psichiatrica e i disturbi d’ansia. Recentemente le era stata diagnosticata un’ipertensione moderata e aveva avuto difficoltà a rimanere incinta negli ultimi anni con 3 aborti consecutivi nel primo trimestre. I sintomi neurologici includevano neuropatia che coinvolgeva i piedi, parestesia intermittente delle gambe e una storia di emicrania cronica per diversi mesi.
L’esame dermatologico ha rivelato una donna leggermente sovrappeso con un motivo scuro, eritematoso, irregolare, netto di 25×30 cm sul lato sinistro del tronco superiore e inferiore (Figura 1). Livedo racemosa esteso non è stato alterato dai cambiamenti di temperatura ed è rimasto invariato per più di 9 mesi. Non c’erano segni di prurito o ulcerazioni e le aree di livedo racemosa erano leggermente tenere alla palpazione.
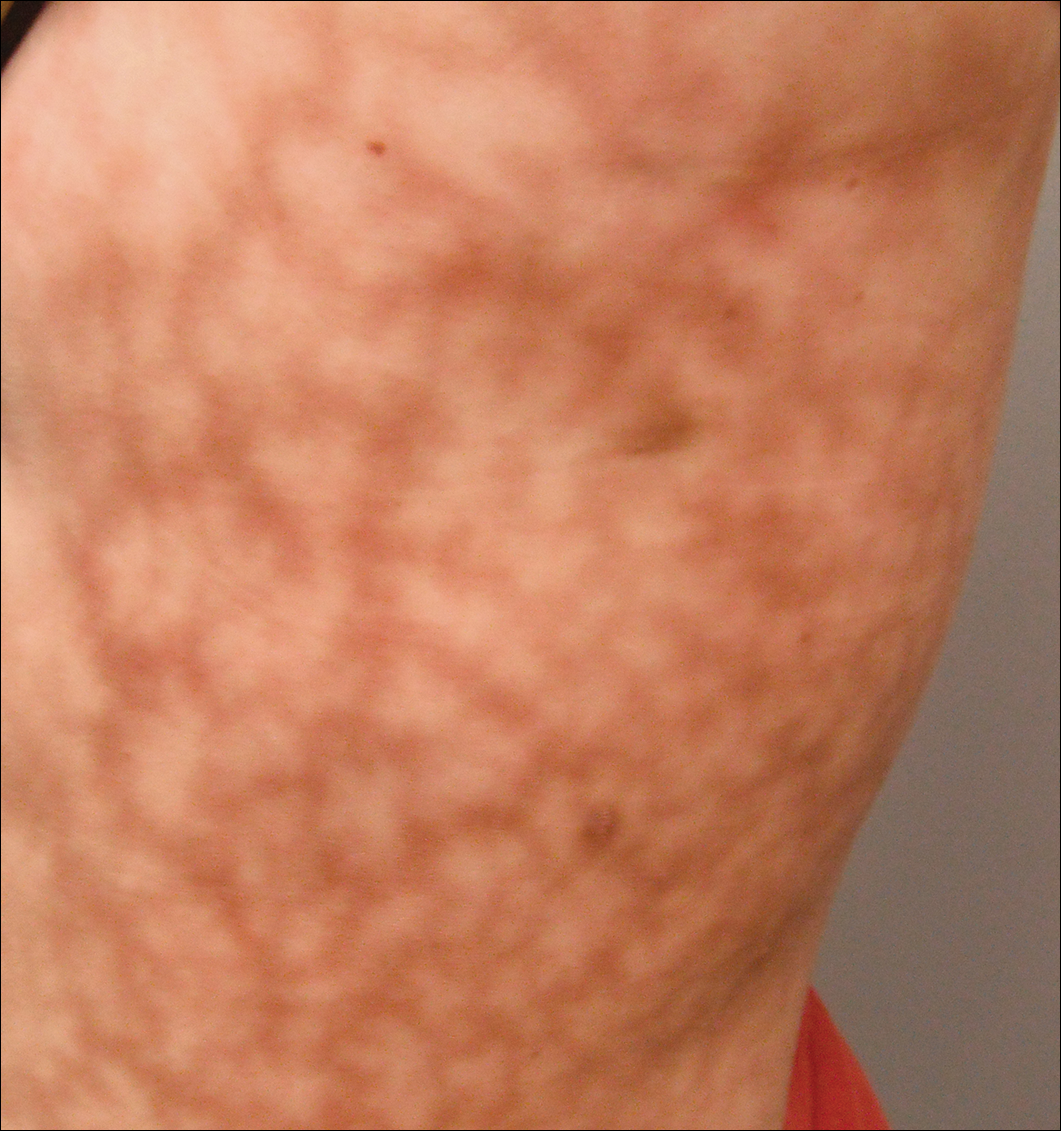
Figura 1. Livedo racemosa si presenta come un motivo violaceo a rete sul lato sinistro del tronco di 25×30 cm.
Abbiamo eseguito 2 serie di tre biopsie da 4 mm. Il primo set aree mirate all’interno del modello violaceo, mentre il secondo set aree mirate di tessuto normale tra le aree screziate. Tutti e 6 i campioni hanno dimostrato infiltrato linfocitario perivascolare superficiale senza evidenza di vasculite o malattia del tessuto connettivo. I vasi non hanno mostrato microtrombi o fibrosi circostante. Nessun eosinofilo è stato identificato all’interno dell’epidermide. Non c’era evidenza di aumento della mucina dermica. Sia i plessi vascolari superficiali che profondi erano insignificanti e non mostravano alcuna prova di danni alle pareti (Figura 2).
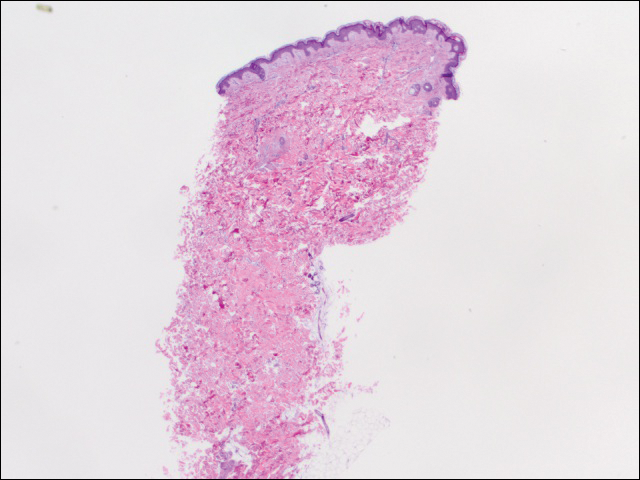
Figura 2. La biopsia del punzone dal lato sinistro del tronco ha mostrato melanoderma focale e infiltrato linfocitico superficiale perivascolare sparso senza evidenza di vasculite, microtrombi o deposizione di fibrina (H&E, ingrandimento originale ×20).
Per escludere altre possibili cause di livedo racemosa, sono stati eseguiti il conteggio completo delle cellule del sangue, il pannello metabolico completo, il profilo di coagulazione, il test della lipasi, l’analisi delle urine, il test sierologico e il workup immunologico. La lipasi era nell’intervallo di riferimento. Il conteggio completo delle cellule del sangue ha rivelato anemia lieve, mentre il resto dei valori erano all’interno dell’intervallo di riferimento. Un workup immunologico includeva l’antigene A della sindrome di Sjögren, l’antigene B della sindrome di Sjögren, gli anticorpi anticardiolipina e l’anticorpo antinucleare, che erano tutti negativi. La storia familiare era notevole per i parenti di primo grado con lupus eritematoso sistemico e malattia di Crohn.
La tomografia computerizzata ha rivelato l’allargamento della milza, così come la linfoadenopatia periaortica, portacavale e porta hepatis. Sulla base dei risultati di laboratorio e della presentazione clinica, nonché dell’anamnesi del paziente, la diagnosi di esclusione è stata livedo racemosa idiopatica con progressione sconosciuta a SS in piena regola. Il paziente non ha soddisfatto gli attuali criteri diagnostici per SS, e i suoi studi immunologici non sono riusciti a confermare eventuali anticorpi presenti, ma il coinvolgimento del sistema reticoloendoteliale ha indicato la produzione di anticorpi che non erano ancora rilevabili sui test di laboratorio.