Ważne ogłoszenie
amyloidoza to grupa chorób, które mają wspólną cechę odkładania fibrilu amyloidu w różnych narządach i tkankach. Układowe typy amyloidozy są bardzo różne od siebie w odniesieniu do biochemicznego charakteru złogów amyloidu i prekursora białka amyloidu. Niektóre są nabywane, a inne dziedziczone. Niektóre typy dzielą kliniczne cechy choroby w odniesieniu do zaangażowania narządów i dysfunkcji. Kluczowe jest zdefiniowanie prekursora białka amyloidogennego i dokładne typowanie złogów amyloidu, aby móc zaoferować dokładne leczenie, ocenić rokowanie i zasugerować poradnictwo genetyczne w razie potrzeby.
amyloidoza AL (pierwotna)
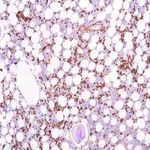
al (immunoglobulin light chain) amyloidoza jest nabytym zaburzeniem komórek plazmatycznych, w którym monoklonalny łańcuch lekki immunoglobulin jest wytwarzany w szpiku kostnym i zwykle znajduje się we krwi lub moczu. Amyloidoza al czasami występuje ze szpiczakiem mnogim, chłoniakiem lub makroglobulinemią Waldenstorma. Fibryle amyloidowe w tego typu amyloidozie składają się z białek łańcucha lekkiego immunoglobuliny (kappa lub lambda). Objawy mogą wystąpić w każdym narządzie organizmu i obejmują niewydolność serca, białko w moczu lub niewydolność nerek, powiększenie wątroby, neuropatię lub powiększony język. Leczenie chemioterapią, w tym chemioterapią w wysokich dawkach z przeszczepem komórek macierzystych i innymi środkami, może doprowadzić do remisji choroby szpiku kostnego, co prowadzi do poprawy dysfunkcji narządów i ochrony narządów docelowych. Jest to jeden z najczęstszych rodzajów amyloidozy zdiagnozowano w Stanach Zjednoczonych.
AA amyloidoza (wtórna)
AA amyloidoza jest spowodowana przewlekłym zakażeniem lub chorobą zapalną, taką jak reumatoidalne zapalenie stawów, rodzinna gorączka śródziemnomorska (FMF), zapalenie szpiku lub ziarniniakowe zapalenie jelita krętego. Infekcja lub stan zapalny powoduje podwyższenie białka ostrej fazy, SAA, z których część złogów jako włókien amyloidowych. Dlatego jest określany jako amyloidoza AA. AA amyloidoza zwykle zaczyna się jako choroba w nerkach, ale inne narządy mogą mieć wpływ. Medyczne lub chirurgiczne leczenie podstawowej przewlekłej infekcji lub choroby zapalnej może spowolnić lub zatrzymać postęp tego typu amyloidozy.
ATTR amyloidosis
ATTR amyloidosis (ATTRm or ATTRwt)
There are several types of inherited amyloidoses, the most common of which is caused by a mutation in the transthyretin (TTR) gene that produces abnormal transthyretin protein. The abnormal TTR protein deposits as amyloid fibrils: Hence, it is termed ATTR amyloidosis. Objawy choroby są zwykle neuropatii i kardiomiopatii i występują w połowie do późnego życia. ATTR amyloidoza znajduje się w rodzinach prawie każdego pochodzenia etnicznego. Znanych jest ponad 100 różnych mutacji transtyretyny i większość powoduje amyloidozę. ATTR może również wystąpić z dzikim typem niezmutowanego białka TTR, zwykle powodując kardiomiopatię u starszych mężczyzn; wcześniej był określany jako „starcza układowa amyloidoza”, ale teraz jest znany jako amyloidoza ATTRwt. Opcje leczenia obejmują stabilizatory TTR (Diflunisal lub Tafamidis) lub tłumiki genów lub produkcję TTR lub przeszczep wątroby.
Inne dziedziczne amyloidozy
istnieją inne mutacje genowe, które produkują białka, które powodują amyloidozę. Są one bardzo rzadkie. Należą do nich apolipoproteina A-I (AApoAI), apolipoproteina a-II (aapoaii), gelsolina (AGel), fibrynogen (AFib) i lizozym (ALys). Białko znane jako Lect2 może również powodować amyloidozę; nie jest jasne, czy jest to dziedziczone, czy nie.
Beta-2 Mikroglobulina amyloidoza (Abeta2m)
Beta-2 mikroglobulina amyloidoza jest spowodowana przewlekłą niewydolnością nerek i często występuje u pacjentów dializowanych przez wiele lat. Złogi amyloidu są wykonane z białka beta-2 mikroglobuliny, które gromadzą się w tkankach, szczególnie wokół stawów, gdy nie mogą być wydalane przez nerki z powodu niewydolności nerek.
zlokalizowane amyloidozy (ALoc)
istnieje wiele rodzajów zlokalizowanych amyloidozy. Zlokalizowane złogi amyloidu w drogach oddechowych (tchawicy lub oskrzeli), oka lub pęcherza moczowego są często spowodowane przez lokalną produkcję łańcuchów lekkich immunoglobulin, nie pochodzących ze szpiku kostnego. Zlokalizowane AL mogą być leczone radioterapią, i to i inne zlokalizowane formy czasami są podatne na operację. Inne zlokalizowane rodzaje amyloidozy są związane z białek endokrynologicznych, lub białka produkowane w skórze, serce, i innych miejscach. Zwykle nie stają się one ogólnoustrojowe.