Linfócitos Granulares grandes Leucemia como uma Complicação da Artrite reumatóide | Reumatología Clínica
Introdução
a síndrome de Felty (FS) é uma rara complicação sistêmica (menos de 1%) de artrite reumatóide (ar), caracterizada pela tríade de RA, a neutropenia persistente (3
) e esplenomegalia de tamanho variável, que pode variar de subclínica esplenomegalia, só detectáveis por imagem, para massive1 esplenomegalia. Ocorre principalmente em casos de longa data com doença articular grave e manifestações extraarticulares, e tem uma forte associação com o haplótipo HLA-DR4 (quase 95% dos casos).2 em 30% -40% dos doentes com FS existe uma expansão dos linfócitos granulares grandes (LGL).1 LGL representa 10% -15% das células mononucleares circulantes e são morfologicamente identificadas pelo seu grande tamanho (15–18µm), seu núcleo redondo ou indentado e citoplasma abundante com grânulos azurofílicos. O fenótipo destas células pode ser linfócito T citotóxico (CD8+, CD57+) ou matador natural (NK) (CD3−, CD8−, CD56+).3 Quando a expansão da LGL é monoclonal e está associada a infiltração da medula óssea e do baço por estas células, é denominada leucemia de linfócitos granulares grandes (LGLL) e é considerada uma doença linfoproliferativa crónica de baixo grau. Sua apresentação clínica é semelhante à de FS, destacando a maior susceptibilidade a infecções bacterianas associadas com neutropenia, anemia e esplenomegalia, como também tem sido chamado de “pseudo-Felty”.3,4 Clinical Presentation
a 70-year-old retired worker from a granite quarry was diagnosticed at age 43 with seropositive RA with involvement of hands, feet, knees and hips. Posteriormente desenvolveu pneumoconiose e nódulos reumatóides pulmonares (Fig. 1), sendo diagnosticado como síndrome de Caplan. Durante a sua progressão, foi tratado com AINEs, corticosteróides, sais de ouro, ciclosporina e metotrexato. Apesar disso, o paciente desenvolveu danos estruturais nas mãos, pés e quadris, exigindo a colocação de próteses em ambos os quadris aos 51 e 54 anos de idade, respectivamente. Nos últimos anos, a sua doença manteve-se estável, tratada com metotrexato 10 mg por semana e doses baixas de glucocorticóides, sem evidência de actividade inflamatória articular. Ele apresentou deformidades de “gooseneck” em todos os dedos e nódulos reumatóides nos cotovelos. Há 12 meses, desenvolveu subitamente febre e dor na virilha direita, com subsequente choque séptico, demonstrando uma infecção da anca direita por Salmonella spp. Metotrexato e glucocorticóides foram suspensos, e ele foi tratado com terapia antibiótica prolongada e substituição parcial da prótese. Os testes laboratoriais revelaram neutropenia persistente, apesar da interrupção dos fármacos mielotóxicos e da melhoria da sépsis, atingindo 0neutrofilos/mm3. Retrospectivamente, ao rever os números de neutrófilos, ele teve contagens baixas um ano antes, entre 1800 e 1000/mm3. O resto da contagem de sangue e bioquímica estavam normais. O ESR foi de 80 mm/h E o CRP 111mg/l. manteve níveis elevados de factor reumatóide (6930U/ml) e anti-CCP (300U/ml) e também teve hipergamaglobulinemia policlonal. Os anticorpos antinucleares e os antigénios nucleares extraíveis eram negativos e os níveis de complemento estavam dentro dos valores normais. A tipagem HLA mostrou que ele carregava o haplótipo DRB1 * 0404 (DR4) e esplenomegalia leve foi detectada (13,7 cm) na tomografia computadorizada abdominal.

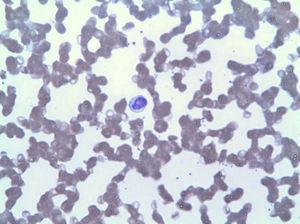
O esfregaço de sangue periférico mostrou linfocitose por LGL (Fig. 2), que no imunofenótipo correspondia a 42% dos leucócitos totais, com um fenótipo aberrante do linfócito T citotóxico (CD3+, CD8+, CD5+, CD7+/−, CD4−, CD56− e DR+). A biópsia da medula óssea mostrou 20% da celularidade total da medula correspondente à mesma expansão clonal (confirmada pelo rearranjo da região variável de TCR gama). Com base nestes resultados, foi feito um diagnóstico de LGLL e o tratamento com metotrexato de 15 mg por semana reiniciado apesar de 3 meses com neutropenia persistente (3
), requerendo a administração frequente de factores estimuladores da granulopoiese. Foi subsequentemente tratado com ciclofosfamida, vincristina e prednisona em doses elevadas. Após 6 meses de tratamento, a neutropenia persiste.
Discussão
LGLL é uma leucemia crônica, caracterizada pela expansão da LGL monoclonais fenótipo ativado linfócitos T ou, menos freqüentemente, as células NK.5 a idade média de diagnóstico é de 60 anos e é frequentemente associada a doenças auto-imunes, particularmente ar, mas também tem sido descrita em colite ulcerativa, síndrome de Sjögren, lúpus eritematoso e esclerose multiple1. Os doentes com ar que tenham associado LGLL têm uma apresentação clínica semelhante à da SF. São geralmente doentes com ar de longa data, lesões articulares graves e consequências significativas, e aumento da frequência de manifestações extra-articulares tais como nódulos reumatóides, linfadenopatia, úlceras pré-bacterianas, pleurite, pigmentação cutânea, neuropatia ou episclerite.6 O paciente teve outra complicação rara da RA, pneumoconiose reumatóide ou Caplan síndrome, caracterizada pelo aparecimento de nódulos pulmonares com o exame histopatológico semelhante ao típica de nódulos reumatóides em pacientes com história de exposição ocupacional a poeiras inorgânicas, tais como sílica, carvão ou granito.7 tanto quanto sabemos, o presente caso é o primeiro que descreve a apresentação da síndrome de Caplan e da LGLL no mesmo paciente. Na maioria dos casos, a lgll de apresentação é neutropenia grave associada a infecções bacterianas recorrentes. Os microrganismos mais frequentemente envolvidos são Staphylococcus aureus, Streptococcus spp. e bacilos gram-negativos. Menos comum anemia, febre, suores nocturnos e aumento do fígado e do baço5 também pode acompanhá-lo. Até um terço dos doentes com LLGG não têm actividade clínica aparente de ar no momento do diagnóstico, mas mantêm níveis elevados de RSE.6 até 40% dos doentes com SF têm linfocitose LGL.7 este fato, junto com a semelhança clínica e associação com HLA-DR4 tem sugerido fortemente que FS e LGLL associados com RA são expressões da mesma entidade caracterizada pela proliferação de LGL.Outras formas incluem também formas mais leves, tais como linfocitose reativa e infecções para formas mais agressivas de LGLL NK5. O diagnóstico LGLL é baseado na descoberta de uma expansão monoclonal da LGL no sangue periférico e na medula óssea com um imunofenótipo característico (CD3+, CD4−, CD8 +, CD16+, CD28− e CD57+). A clonalidade é confirmada através do estudo do gene reTCR9. Em geral, a LGL tem uma progressão crônica e indolente, com uma sobrevivência média de 10 anos.1 em casos raros, especialmente quando a expansão é devido à LGL com fenótipo NK, esta leucemia pode se comportar de forma mais agressiva.A indicação mais comum para o tratamento são infecções recorrentes e, menos frequentemente, anemia, esplenomegalia sintomática ou o aparecimento de sintomas B B1 graves.
o tratamento de primeira linha na LGLL é imunossupressor isolado, especificamente metotrexato (10 mg / semana), ciclosporina a (1–1.5 mg/kg/2 vezes por dia) ou ciclofosfamida oral (50-100 mg/dia). Este tratamento é eficaz em cerca de 50% dos doentes, alcançando a correcção das citopenias, mas não erradicando as células leucémicas.1 glucocorticóides podem ser utilizados para acelerar a resposta e os factores estimuladores da granulopoiese são úteis no tratamento inicial da neutropenia. Em doentes refractários e em doentes com apresentação muito agressiva, foi tentado o tratamento com regimes de quimioterapia semelhantes ao CHOP (ciclofosfamida, vincristina, doxorrubicina e prednisona) e outros regimes para o linfoma, mas não demonstraram claramente a sua eficácia. Outros tratamentos que foram testados são análogos da purina, Alemtuzumab, bortezomib, esplenectomia e transplante alogénico de medula óssea, com resultados variáveis.3 conclusões tanto FS como LGLL são complicações raras da AR, que aparecem em doenças de longa data, com danos estruturais significativos e manifestações extra-articulares. Em doentes com ar de longa duração e neutropenia devem ser excluídas a presença de proliferações clonais de LGL no sangue periférico e/ou na medula óssea, permitindo o diagnóstico de LGL. O tratamento de primeira linha é o uso de fármacos imunossupressores, tais como metotrexato em dose baixa, e os glucocorticóides podem estar associados a factores estimuladores da granulopoiese. Outras modalidades de tratamento, tais como quimioterapia ou esplenectomia, têm mostrado resultados variáveis em alguns casos refratários.divulgações éticas protecção dos seres humanos e dos animais. Os autores declaram que não foram realizadas experiências em humanos ou animais para esta investigação.confidencialidade dos dados. Os autores declaram ter seguido os protocolos de seu centro de trabalho sobre a publicação de dados de pacientes e que todos os pacientes incluídos no estudo receberam informações suficientes e deram o seu consentimento informado por escrito para participar nesse estudo.