Leucemia limfocitară granulară mare ca o complicație a artritei reumatoide / reumatolog Ecqua Cl Ecquxnica
introducere
sindromul Felty (FS) este o complicație sistemică rară (mai puțin de 1%) a artritei reumatoide (RA), caracterizată prin triada RA, neutropenie persistentă (3
) și splenomegalie de dimensiuni variabile, care poate varia de la splenomegalie subclinică, detectabilă numai prin imagistică, până la splenomegalie masivă1. Apare în principal în cazuri de lungă durată cu boală articulară severă și manifestări extraarticulare și are o asociere puternică cu haplotipul HLA-DR4 (aproape 95% din cazuri).2 la 30% -40% dintre pacienții cu FS există o expansiune a limfocitelor granulare mari (LGL).1 LGL reprezintă 10% -15% din celulele mononucleare circulante și sunt identificate morfologic prin dimensiunea lor mare (15–18 MMC), nucleul lor rotund sau indentat și citoplasma abundentă cu granule azurofile. Fenotipul acestor celule poate fi limfocite T citotoxice (CD8+, CD57+) sau killer natural (NK) (CD3−, CD8−, CD56+).3 când extinderea LGL este monoclonală și este asociată cu infiltrarea măduvei osoase și a splinei de către aceste celule, se numește leucemie limfocitară granulară mare (LGLL) și este considerată o tulburare limfoproliferativă cronică de grad scăzut. Prezentarea sa clinică este similară cu cea a FS, evidențiind susceptibilitatea crescută la infecții bacteriene asociate cu neutropenie, anemie și splenomegalie, așa cum a fost numită și „pseudo-Felty”.3,4 prezentare clinică
un muncitor pensionar de 70 de ani dintr-o carieră de granit a fost diagnosticat la vârsta de 43 de ani cu RA seropozitiv cu implicarea mâinilor, picioarelor, genunchilor și șoldurilor. Ulterior a dezvoltat pneumoconioză și noduli reumatoizi pulmonari (Fig. 1), fiind diagnosticat ca sindromul Caplan. În timpul progresiei sale, a fost tratat cu AINS, corticosteroizi, săruri de aur, ciclosporină și metotrexat. În ciuda acestui fapt, pacientul a dezvoltat leziuni structurale la nivelul mâinilor, picioarelor și șoldurilor, necesitând plasarea protezei în ambele șolduri la vârsta de 51 și respectiv 54 de ani. În ultimii ani, boala sa a rămas stabilă, tratată cu metotrexat 10 mg săptămânal și doze mici de glucocorticoizi, fără dovezi de activitate articulară inflamatorie. El a prezentat deformări ale gâtului de gâscă în toate degetele și noduli reumatoizi pe coate. În urmă cu 12 luni, el a dezvoltat brusc febră și durere în zona inghinală dreaptă, cu șoc septic ulterior, demonstrând o infecție a șoldului drept de Salmonella spp. Metotrexatul și glucocorticoizii au fost suspendați și a fost tratat cu terapie antibiotică prelungită și înlocuirea parțială a protezei. Testele de laborator au evidențiat neutropenie persistentă, în ciuda retragerii medicamentelor mielotoxice și îmbunătățirea sepsisului, ajungând la 0neutrofile/mm3. Retrospectiv, la revizuirea numărului de neutrofile, el a avut un număr scăzut cu un an mai devreme, între 1800 și 1000/mm3. Restul numărului de sânge și biochimie au fost normale. ESR a fost de 80mm/h și CRP 111mg/l. el a menținut niveluri ridicate de factor reumatoid (6930u/ml) și anti-CCP (300U / ml) și a avut, de asemenea, hipergamaglobulinemie policlonală. Anticorpii antinucleari și antigenii nucleari extractibili au fost negativi, iar nivelurile complementului s-au încadrat în valori normale. Tastarea HLA a arătat că a purtat haplotipul DRB1*0404 (DR4) și a fost detectată splenomegalie ușoară (13,7 cm) pe tomografia computerizată abdominală.

pneumoconioză și noduli reumatoizi pulmonari.
frotiul de sânge periferic a arătat limfocitoză prin LGL (Fig. 2), care în imunofenotip corespundea la 42% din totalul leucocitelor, cu un fenotip aberant al limfocitelor T citotoxice (CD3+, CD8+, CD5+, CD7+/−, CD4−, CD56− și DR+). Biopsia măduvei osoase a arătat 20% din celularitatea totală a măduvei corespunzătoare aceleiași expansiuni clonale (confirmată prin rearanjarea regiunii variabile a TCR gamma). Pe baza acestor constatări, a fost pus un diagnostic de LGLL și tratamentul cu metotrexat 15 mg săptămânal a fost reluat în ciuda a 3 luni cu neutropenie persistentă (3
), necesitând administrarea frecventă a factorilor stimulatori ai granulopoiezei. Ulterior a fost tratat cu ciclofosfamidă, vincristină și prednison la doze mari. După 6 luni de tratament, neutropenia persistă.
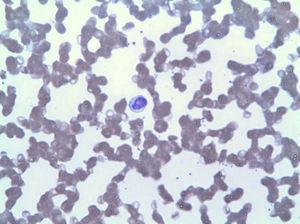
limfocitoză datorată LGL.
discuție
LGLL este o leucemie cronică caracterizată prin extinderea fenotipului monoclonal LGL al limfocitelor T citotoxice activate sau a celulelor NK mai puțin frecvent.5 vârsta medie a diagnosticului este de 60 de ani și este frecvent asociată cu boli autoimune, în special RA, dar a fost descrisă și în colita ulcerativă, sindromul SJ, lupus eritematos și scleroză multiple1. Pacienții cu PR care au asociat LGLL au o prezentare clinică similară cu cea a FS. Acestea sunt de obicei pacienți cu RA de lungă durată, leziuni articulare severe și consecințe semnificative și frecvență crescută a manifestărilor extraarticulare, cum ar fi noduli reumatoizi, limfadenopatie, ulcere pretibiale, pleurită, pigmentare a pielii, neuropatie sau episclerită.6 pacientul a avut, de asemenea, o altă complicație rară a RA, pneumoconioza reumatoidă sau sindromul Caplan, caracterizată prin apariția nodulilor pulmonari cu histopatologie similară cu cea a nodulilor reumatoizi tipici la pacienții cu antecedente de expunere profesională la pulberi anorganice precum silice, cărbune sau granit.7 din câte știm, cazul de față este primul care descrie prezentarea sindromului Caplan și a LGLL la același pacient. În cele mai multe cazuri, prezentarea LGLL este neutropenie severă asociată cu infecții bacteriene recurente. Microorganismele cele mai frecvent implicate sunt Staphylococcus aureus, Streptococcus spp. și bacili gram-negativi. Mai puțin frecvent anemia, febra, transpirațiile nocturne și mărirea ficatului și a splinei5 îl pot însoți, de asemenea. Până la o treime dintre pacienții cu LLGG nu au activitate clinică par aparentă la momentul diagnosticului, dar mențin niveluri ridicate de ESR.6 până la 40% dintre pacienții cu FS au limfocitoză LGL.7 acest fapt, împreună cu similitudinea clinică și asocierea cu HLA-DR4, a sugerat cu tărie că FS și LGLL asociate cu RA sunt expresii ale aceleiași entități caracterizate prin proliferarea LGL.8 alte forme includ, de asemenea, forme mai blânde, cum ar fi limfocitoza reactivă și infecțiile la forme mai agresive de NK5 LGLL. Diagnosticul LGLL se bazează pe constatarea unei expansiuni monoclonale a LGL în sângele periferic și măduva osoasă cu un imunofenotip caracteristic (CD3+, CD4−, CD8 +, CD16+, CD28− și CD57+). Clonalitatea este confirmată prin studierea genei reTCR9. În general, LGL are o progresie cronică și indolentă, cu o supraviețuire medie de 10 ani.1 în cazuri rare, mai ales atunci când extinderea se datorează LGL cu fenotip NK, această leucemie se poate comporta mai agresiv.5 cea mai frecventă indicație pentru tratament sunt infecțiile recurente și, mai puțin frecvent, anemia, splenomegalia simptomatică sau apariția simptomelor B severe B1.
tratamentul de primă linie în LGLL sunt medicamente imunosupresoare singure, în special metotrexat (10 mg/săptămână), ciclosporină A (1-1, 5 mg/kg/2 ori pe zi) sau ciclofosfamidă orală (50-100 mg / zi). Acest tratament este eficient la aproximativ 50% dintre pacienți, realizând corectarea citopeniilor, dar nu eradicând celulele leucemice.1 glucocorticoizii pot fi utilizați pentru a accelera răspunsul, iar factorii de stimulare a granulopoiezei sunt utili în tratamentul inițial al neutropeniei. La pacienții refractari și la cei cu tratament de prezentare foarte agresiv cu regimuri de chimioterapie similare cu CHOP (ciclofosfamidă, vincristină, doxorubicină și prednison) și alte scheme de limfom au fost încercate, dar nu și-au demonstrat clar eficacitatea. Alte tratamente care au fost testate sunt analogii purinici, Alemtuzumab, bortezomib, splenectomia și transplantul de măduvă osoasă alogenă, cu rezultate variabile.3
concluzii
atât FS, cât și LSLL sunt complicații rare ale RA, care apar în boala de lungă durată, cu leziuni structurale semnificative și manifestări extraarticulare. La pacienții cu RA și neutropenie de lungă durată, trebuie exclusă prezența proliferărilor clonale ale LGL în sângele periferic și/sau măduva osoasă, permițând diagnosticarea LGLL. Tratamentul de primă linie este utilizarea de medicamente imunosupresoare, cum ar fi metotrexatul cu doze mici, iar glucocorticoizii pot fi asociați cu factori de stimulare a granulopoiezei. Alte modalități de tratament, cum ar fi chimioterapia sau splenectomia, au arătat rezultate variabile în unele cazuri refractare.
dezvăluiri etice
protecția subiecților umani și animali. Autorii declară că nu au fost efectuate experimente pe oameni sau animale pentru această investigație.
confidențialitatea datelor. Autorii declară că au urmat protocoalele Centrului lor de lucru privind publicarea datelor despre pacienți și că toți pacienții incluși în studiu au primit suficiente informații și și-au dat consimțământul în cunoștință de cauză în scris pentru a participa la studiul respectiv.