Idiopatisk Livedo Racemosa Med Splenomegali og Diffus Lymfadenopati
Sneddon syndrom (SS) ble først beskrevet i 1965 hos pasienter med vedvarende livedo racemosa og nevrologiske hendelser.1 fordi DE andre manifestasjoner AV SS er uspesifikke (f. eks hypertensjon, hjerteventulopati, arteriell og venøs okklusjon), blir diagnosen ofte forsinket. Mange pasienter som opplever prodromale nevrologiske symptomer som hodepine, depresjon, angst, svimmelhet og nevropati, presenterer ofte en lege før de utvikler iskemiske hjernemanifestasjoner2, men får sjelden riktig diagnose. Utbruddet av cerebrale okklusive hendelser forekommer vanligvis hos pasienter yngre enn 45 år og kan oppstå som et forbigående iskemisk angrep, slag eller intrakranial blødning.3 sykdommen er mer utbredt hos kvinner enn menn (2: 1-forhold). Den nøyaktige patogenesen AV SS er fortsatt ukjent, og selv om DET har vært tenkt som en separat enhet fra systemisk lupus erythematosus og andre antifosfolipidforstyrrelser, har det blitt postulert at en immunologisk dysfunksjon skader karvegger som fører til trombose.
Kutane funn assosiert MED SS involverer små til mellomstore dermale subdermale arterier. Histopatologi hos noen pasienter viser proliferasjon av endotel – og fibrinavsetninger med påfølgende utslettelse av involverte arterier.4 hos mange pasienter, inkludert vår pasient, viser histopatologisk undersøkelse av involvert hud ikke spesifikke abnormiteter.1 Zelger et al5 rapporterte sekvensen av histopatologiske hudhendelser i en serie antifosfolipidnegative SS-pasienter. Forfatterne rapporterte at bare små arterier ved dermis-subcutis-krysset var involvert, og en progresjon av endotelial dysfunksjon ble observert. Forfatterne trodde det var flere uspesifikke stadier før fibrin okklusjon av involverte arterier.5 Trinn i involvert løsning av endotelceller med uspesifikk perivaskulær lymfocytisk infiltrasjon med perivaskulær betennelse og lymfocytisk infiltrasjon som representerer den primære mover av sykdommen.5,6 dette stadiet antas å være kortvarig, og dermed grunnen til at DET har gått uoppdaget i MANGE år hos SS-pasienter. Trinn II TIL IV fremgang gjennom fibrin avsetning og okklusjon.5 Histologiske trekk i trinn i TIL II er ikke rapportert på grunn av sen DIAGNOSE AV SS. Stage i pasienter vanligvis til stede med en gjennomsnittlig varighet av symptomer på 6 måneder med få nevrologiske symptomer, den vanligste er parestesi i bena.5
Saksrapport
en 37 år gammel kvinne med epigastrisk ømhet på venstre side og splenomegali sett på computertomografi ble henvist av en hematolog for evaluering av et retikulært utslett på venstre side av flanken av 9 måneders varighet med en antatt diagnose av fokal melanoderma. Hennes medisinske historie var bemerkelsesverdig for en medfødt ventrikkelseptumdefekt og koarktasjon, samt endometriose, myalgi og leddstivhet som alle hadde utviklet seg i løpet av det siste året. Hennes medisinske historie var også bemerkelsesverdig for nephrolithiasis, irritabel tarmsyndrom og kronisk bihulebetennelse, samt psykiatrisk depresjon og angstlidelser. Hun hadde nylig blitt diagnostisert med moderat hypertensjon og hadde opplevd problemer med å bli gravid de siste årene med 3 påfølgende miskramper i første trimester. Neurologiske symptomer inkluderte nevropati som involverer føttene, intermitterende parestesi i bena, og en historie med kronisk migrene hodepine i flere måneder.
Dermatologisk undersøkelse viste en litt overvektig kvinne med et 25×30 cm mørkt, erytematøst, uregelmessig, nettlignende mønster på venstre side av øvre og nedre stamme (Figur 1). Omfattende livedo racemosa ble ikke endret av temperaturendringer og hadde vært uendret i mer enn 9 måneder. Det var ingen tegn på kløe eller sårdannelse, og områder med livedo racemosa var litt ømme på palpasjon.
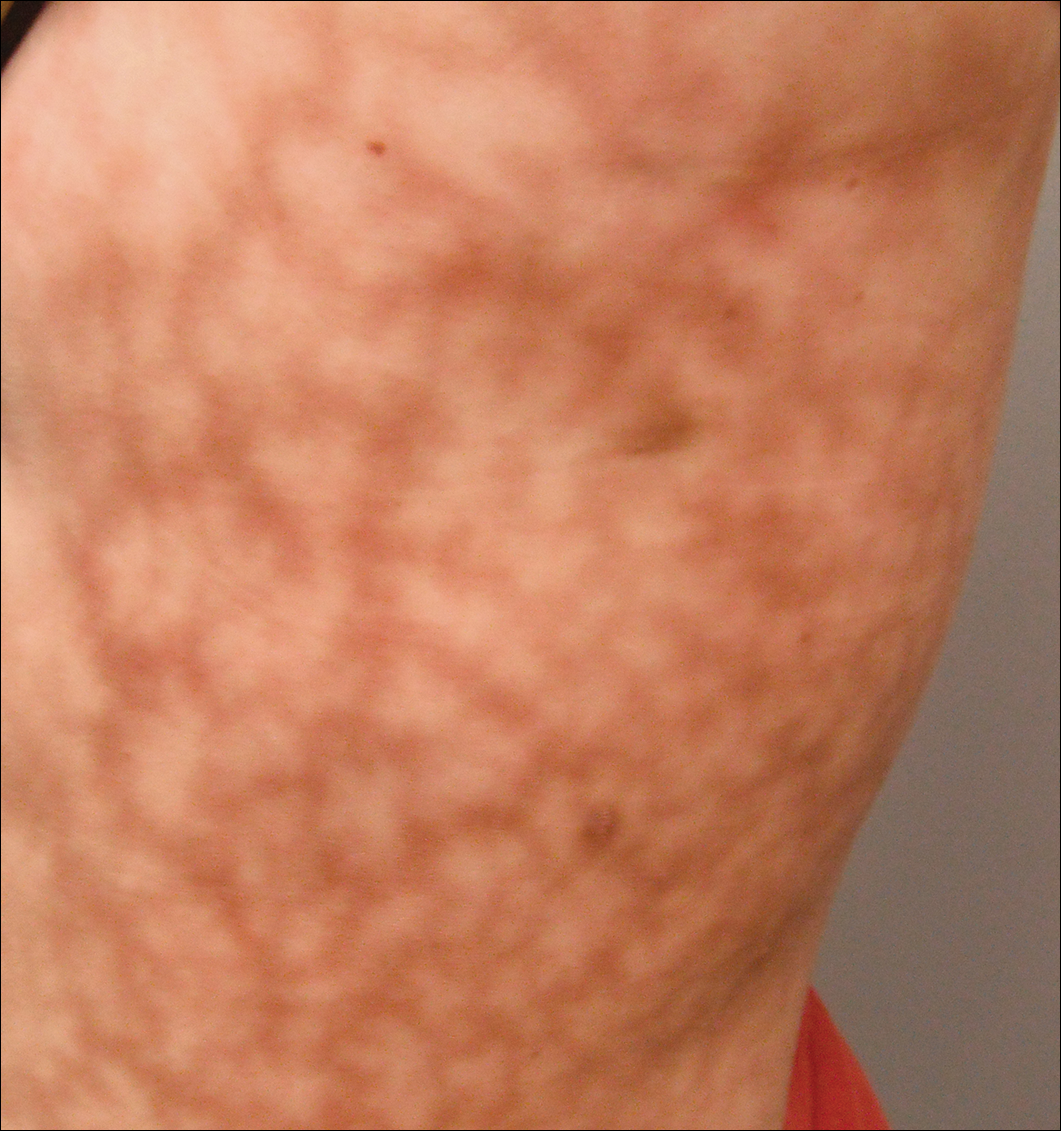
Figur 1. Livedo racemosa presenterer som et nettlignende violaceous mønster på venstre side av stammen som måler 25×30 cm.
vi utførte 2 sett med tre 4 mm biopsier. Det første settet målrettet områder innenfor violaceous mønster, mens det andre settet målrettet områder av normalt vev mellom spraglete områder. Alle 6 prøvene viste overfladisk perivaskulær lymfocytisk infiltrasjon uten tegn på vaskulitt eller bindevevssykdom. Karene viste ingen mikrotrombi eller omkringliggende fibrose. Ingen eosinofiler ble identifisert i epidermis. Det var ingen tegn på økt dermal mucin. Både overfladiske og dype vaskulære plexuser var unremarkable og viste ingen tegn på skade på veggene (Figur 2).
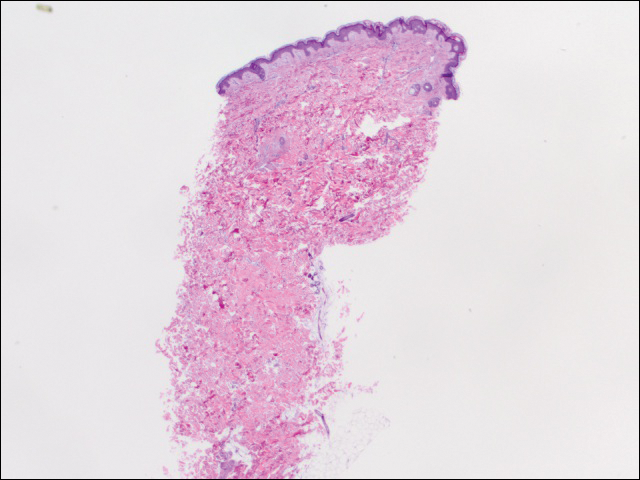
Figur 2. Slagbiopsi fra venstre side av stammen viste fokal melanoderma og sparsom overfladisk perivaskulær lymfocytisk infiltrasjon uten tegn på vaskulitt, mikrotrombi eller fibrinavsetning (H&E, original forstørrelse ×20).
for å utelukke andre mulige årsaker til livedo racemosa, ble det utført komplett blodcelletall, omfattende metabolsk panel, koagulasjonsprofil, lipasetest, urinalyse, serologisk testing og immunologisk undersøkelse. Lipase var innenfor referanseområdet. Det komplette blodcelletallet viste mild anemi, mens resten av verdiene var innenfor referanseområdet. En immunologisk undersøkelse inkluderte Sjö syndromantigen A, Sjö syndromantigen B, antikardiolipinantistoffer og antinukleære antistoff, som alle var negative. Familiehistorie var bemerkelsesverdig for førstegrads slektninger med systemisk lupus erythematosus og Crohns sykdom.
Computertomografi viste forstørrelse av milten, så vel som periaortisk, portacaval og porta hepatis lymfadenopati. Basert på laboratoriefunn og klinisk presentasjon samt pasientens sykehistorie, var eksklusjonsdiagnosen idiopatisk livedo racemosa med ukjent progresjon til FULLVERDIG SS. Pasienten oppfylte ikke de nåværende diagnostiske kriteriene FOR SS, og hennes immunologiske studier klarte ikke å bekrefte noen nåværende antistoffer, men involvering av retikuloendotelialsystemet pekte på produksjon av antistoffer som ennå ikke var detekterbare ved laboratorietesting.