Idiopática Livedo Racemosa Apresentando-se Com Esplenomegalia e Linfadenopatia Difusa
a síndrome de Sneddon (SS) foi descrita pela primeira vez em 1965, em pacientes com persistente livedo racemosa e neurológicas eventos.1 Uma vez que as outras manifestações da SS são não específicas (por exemplo, hipertensão, valvulopatia cardíaca, oclusão arterial e venosa), o diagnóstico é frequentemente atrasado. Muitos pacientes que experimentam sintomas neurológicos prodromais, tais como dores de cabeça, depressão, ansiedade, tonturas e neuropatia, muitas vezes apresentam a um médico antes de desenvolver manifestações cerebrais isquêmicas2, mas raramente recebem o diagnóstico correto. O aparecimento de eventos oclusivos cerebrais ocorre tipicamente em doentes com menos de 45 anos e pode apresentar-se como um acidente isquémico transitório, acidente vascular cerebral ou hemorragia intracraniana.3 a doença é mais prevalente nas fêmeas do que nos machos (razão 2:1). A patogênese exata da SS ainda é desconhecido, e embora tenha sido pensado como uma entidade separada do lúpus eritematoso sistêmico e outras antifosfolípide transtornos, tem sido postulado que uma disfunção imunológica danos as paredes dos vasos, levando a trombose.os achados cutâneos associados à SS envolvem artérias dérmicas de pequeno a médio porte. A histopatologia em alguns doentes demonstra proliferação dos depósitos de endotélio e fibrina com obliteração subsequente das artérias envolvidas.4 em muitos pacientes, incluindo o nosso paciente, o exame histopatológico da pele envolvida não mostra anormalidades específicas.Zelger et al5 notificaram a sequência de acontecimentos histopatológicos cutâneos numa série de doentes com SS antifosfolípidos negativos. Os autores relataram que apenas pequenas artérias na junção derme-subcútis estavam envolvidas e uma progressão da disfunção endotelial foi observada. Os autores acreditavam que havia vários estágios não específicos antes da oclusão fibrina das artérias envolvidas.A fase I envolveu o afrouxamento das células endoteliais com infiltração linfocítica perivascular não específica com inflamação perivascular e infiltração linfocítica representando o motor principal da doença.5,6 pensa-se que esta fase tenha uma duração curta, sendo assim a razão pela qual não foi detectada durante muitos anos em pacientes com SS. As fases II A IV progridem através da deposição fibrina e da oclusão.As características histológicas dos estádios I A II não foram notificadas devido ao diagnóstico tardio de SS. Os doentes em fase I apresentam tipicamente uma duração média de sintomas de 6 meses com poucos sintomas neurológicos, sendo o mais comum a parestesia das pernas.5
relato de Caso
37-year-old mulher com ternura epigástrica no lado esquerdo e esplenomegalia vistos na tomografia computadorizada foi referida por um hematologista para avaliação de uma reticular erupção no lado esquerdo do flanco de 9 meses de duração, com um suposto diagnóstico de focal melanoderma. Sua história médica foi notável por um defeito congênito do septo ventricular e coarctação da aorta, bem como endometriose, mialgia e rigidez articular que tinham se desenvolvido ao longo do último ano. Sua história médica também foi notável para nefrolitíase, síndrome do intestino irritável, sinusite crônica, bem como depressão psiquiátrica e transtornos de ansiedade. Recentemente foi-lhe diagnosticada hipertensão moderada e teve dificuldade em engravidar nos últimos anos com 3 abortos consecutivos no primeiro trimestre. Os sintomas neurológicos incluíram neuropatia envolvendo os pés, parestesia intermitente das pernas, e uma história de enxaquecas crônicas por vários meses.o exame dermatológico revelou uma mulher ligeiramente obesa, com um padrão retorcido de 25×30 cm, eritematoso, irregular, com forma de rede no lado esquerdo do tronco superior e inferior (Figura 1). O extenso livedo racemosa não foi alterado por mudanças de temperatura e permaneceu inalterado por mais de 9 meses. Não houve sinais de prurido ou ulcerações, e áreas de livedo racemosa foram ligeiramente sensíveis à palpação.
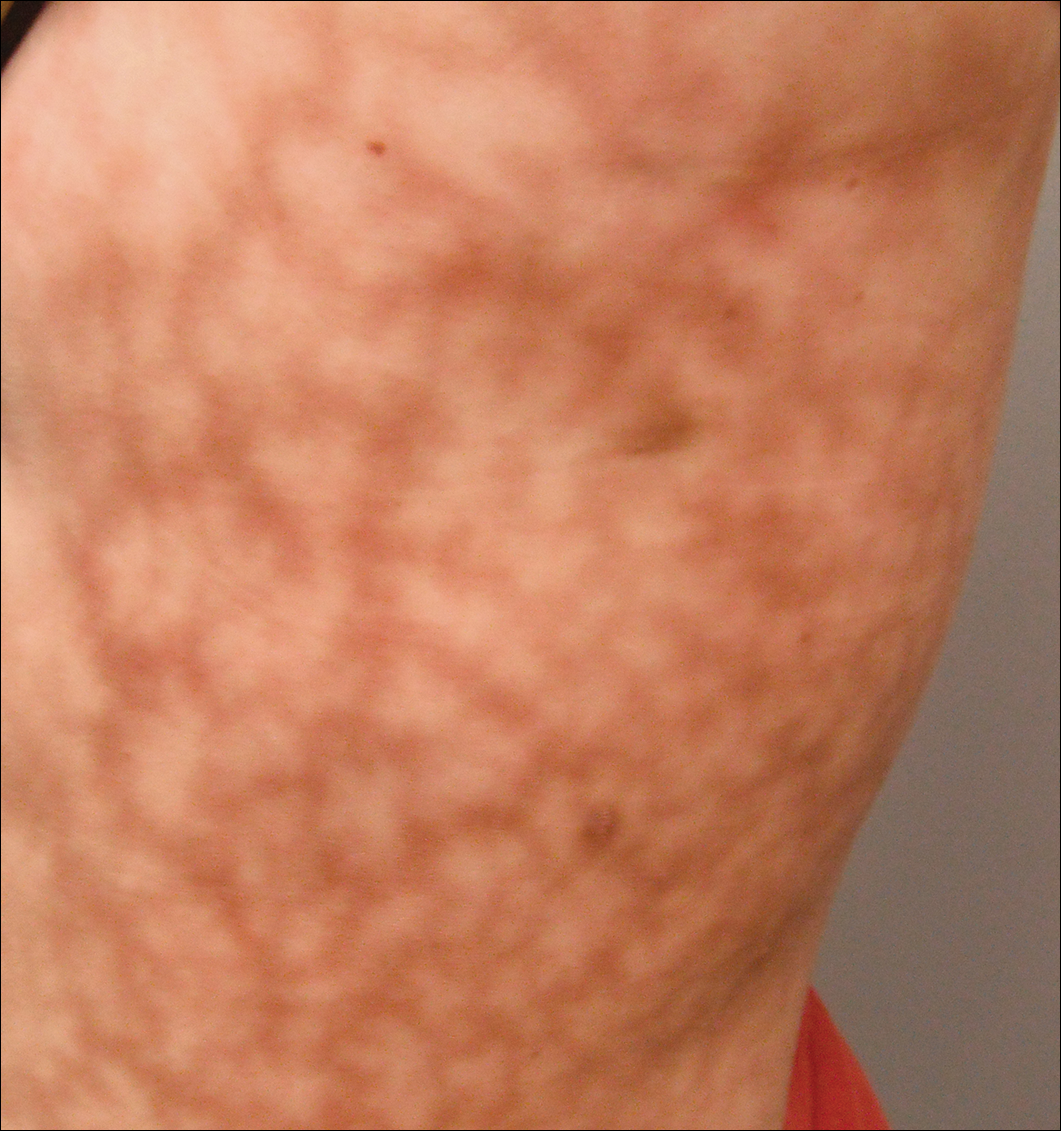
a Figura 1. Livedo racemosa apresenta-se como um padrão violáceo tipo rede no lado esquerdo do tronco medindo 25×30 cm.
realizámos 2 conjuntos de três biópsias de 4 mm. O primeiro conjunto de áreas direcionadas dentro do padrão violáceo, enquanto o segundo conjunto de áreas direcionadas do tecido normal entre as áreas mottled. Todas as 6 amostras demonstraram infiltração linfocítica perivascular superficial sem evidência de vasculite ou doença do tecido conjuntivo. Os vasos não mostraram microtrombi ou fibrose circundante. Não foram identificados eosinófilos na epiderme. Não houve evidência de aumento da mucina dérmica. Tanto os plexos vasculares superficiais como profundos não apresentavam sinais de lesões nas paredes (Figura 2).
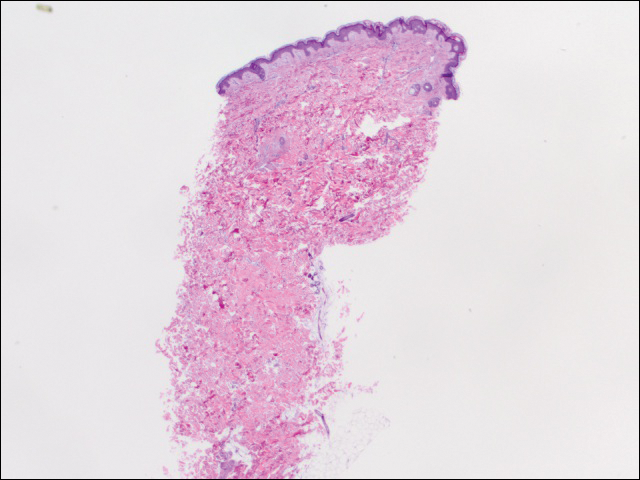
Figura 2. Punção-biópsia do lado esquerdo do tronco mostrou focal melanoderma esparsas e superficial infiltrado linfóide se infiltrar, sem evidência de vasculite, microthrombi, ou deposição de fibrina (H&E, ampliação original ×20).
Para descartar outras possíveis causas de livedo racemosa, hemograma, abrangente metabólica painel de coagulação perfil, lipase de teste, exame de urina, testes sorológicos e imunológicos hemograma foram realizadas. Lipase estava dentro do alcance de referência. A contagem completa de células sanguíneas revelou anemia ligeira, enquanto o resto dos valores estavam dentro do intervalo de referência. Uma análise imunológica incluiu o antigénio a da síndrome de Sjögren, o antigénio B da síndrome de Sjögren, Anticorpos anticardiolipina e anticorpos antinucleares, que foram todos negativos. A história familiar foi notável para parentes de primeiro grau com lúpus eritematoso sistêmico e doença de Crohn.a tomografia computadorizada revelou aumento do baço, assim como periaórticos, portacaval e linfadenopatia porta hepatis. Com base nos resultados laboratoriais e na apresentação clínica, bem como na história médica do paciente, o diagnóstico de exclusão foi livedo racemosa idiopática com progressão desconhecida para SS completa. A doente não preencheu os critérios actuais de diagnóstico para SS, e os seus estudos imunológicos não conseguiram confirmar quaisquer anticorpos presentes, mas o envolvimento do sistema reticuloendotelial apontou para a produção de anticorpos que ainda não foram detectáveis em testes laboratoriais.