Große granuläre Lymphozytenleukämie als Komplikation der rheumatoiden Arthritis / Reumatología Clínica
Einleitung
Das Felty-Syndrom (FS) ist eine seltene systemische Komplikation (weniger als 1%) der rheumatoiden Arthritis (RA), die durch die Trias RA, persistierende Neutropenie (3
) und Splenomegalie unterschiedlicher Größe gekennzeichnet ist, die von subklinischer Splenomegalie, die nur bildgebend nachgewiesen werden kann, bis hin zu massiver1 Splenomegalie reichen kann. Es tritt hauptsächlich in langjährigen Fällen mit schweren Gelenkerkrankungen und extraartikulären Manifestationen auf und hat eine starke Assoziation mit dem HLA-DR4-Haplotyp (fast 95% der Fälle).2 Bei 30% -40% der Patienten mit FS kommt es zu einer Ausdehnung der großen granulären Lymphozyten (LGL).1 LGL repräsentieren 10% -15% der zirkulierenden mononukleären Zellen und sind morphologisch durch ihre große Größe (15–18µm), ihren runden oder eingerückten Kern und reichlich vorhandenes Zytoplasma mit azurophilen Granula identifiziert. Der Phänotyp dieser Zellen kann zytotoxischer T-Lymphozyt (CD8 +, CD57+) oder natürlicher Killer (NK) (CD3−, CD8−, CD56 +) sein.3 Wenn die Expansion von LGL monoklonal ist und mit einer Infiltration des Knochenmarks und der Milz durch diese Zellen einhergeht, spricht man von einer großkörnigen Lymphozytenleukämie (LGLL) und gilt als chronisch niedriggradige lymphoproliferative Erkrankung. Sein klinisches Erscheinungsbild ähnelt dem von FS und unterstreicht die erhöhte Anfälligkeit für bakterielle Infektionen im Zusammenhang mit Neutropenie, Anämie und Splenomegalie, wie sie auch als „Pseudo-Felty“ bezeichnet wird.3,4Klinisches Erscheinungsbild
Bei einem 70-jährigen Rentner aus einem Granitsteinbruch wurde im Alter von 43 Jahren eine seropositive RA mit Beteiligung von Händen, Füßen, Knien und Hüften diagnostiziert. Anschließend entwickelte er Pneumokoniose und Lungenrheumaknoten (Abb. 1), als Caplan-Syndrom diagnostiziert. Während seines Fortschreitens wurde er mit NSAIDs, Kortikosteroiden, Goldsalzen, Cyclosporin und Methotrexat behandelt. Trotzdem entwickelte der Patient strukturelle Schäden an Händen, Füßen und Hüften, die die Platzierung einer Prothese in beiden Hüften im Alter von 51 bzw. 54 Jahren erforderten. In den letzten Jahren blieb seine Krankheit stabil, behandelt mit Methotrexat 10 mg wöchentlich und niedrigen Dosen von Glukokortikoiden, ohne Anzeichen einer entzündlichen Gelenkaktivität. Er präsentierte Schwanenhalsdeformitäten in allen Fingern und Rheumaknoten an den Ellenbogen. vor 12 Monaten entwickelte er plötzlich Fieber und Schmerzen in der rechten Leiste mit anschließendem septischem Schock, was eine Infektion der rechten Hüfte durch Salmonella spp. Methotrexat und Glukokortikoide wurden suspendiert, und er wurde mit einer verlängerten Antibiotikatherapie und einem teilweisen Ersatz der Prothese behandelt. Labortests zeigten eine persistierende Neutropenie, trotz des Entzugs von myelotoxischen Medikamenten und der Verbesserung der Sepsis, erreichen 0neutrophile/mm3. Retrospektiv hatte er bei der Überprüfung der Anzahl der Neutrophilen ein Jahr zuvor niedrige Werte zwischen 1800 und 1000 / mm3. Der Rest des Blutbildes und der Biochemie war normal. Die ESR betrug 80 mm / h und CRP 111 mg / l. Er behielt hohe Spiegel an Rheumafaktor (6930 U / ml) und Anti-CCP (300 U / ml) bei und hatte auch polyklonale Hypergammaglobulinämie. Antinukleäre Antikörper und extrahierbare nukleare Antigene waren negativ und die Komplementspiegel lagen innerhalb normaler Werte. Die HLA-Typisierung zeigte, dass er den Haplotyp DRB1 * 0404 (DR4) trug und eine leichte Splenomegalie (13,7 cm) in der abdominalen Computertomographie nachgewiesen wurde.

Pneumokoniose und Lungenrheumaknoten.
Der periphere Blutausstrich zeigte eine Lymphozytose durch LGL (Abb. 2), die im Immunphänotyp 42% der gesamten Leukozyten entsprach, mit einem aberranten Phänotyp von zytotoxischen T−Lymphozyten (CD3+, CD8+, CD5+, CD7+/−, CD4−, CD56- und DR +). Die Knochenmarkbiopsie zeigte 20% der gesamten Markzellularität, die der gleichen klonalen Expansion entsprach (bestätigt durch Umlagerung der variablen Region von TCR-Gamma). Basierend auf diesen Befunden wurde eine Diagnose von LGLL gestellt und die Behandlung mit Methotrexat 15 mg wöchentlich trotz 3 Monaten mit anhaltender Neutropenie (3
), die eine häufige Verabreichung von Granulopoese-stimulierenden Faktoren erforderte, wieder aufgenommen. Anschließend wurde er in hohen Dosen mit Cyclophosphamid, Vincristin und Prednison behandelt. Nach 6 Monaten Behandlung bleibt die Neutropenie bestehen.
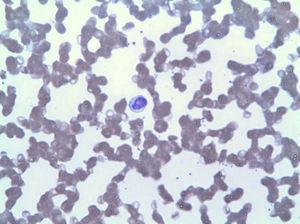
Lymphozytose durch LGL.
Diskussion
LGLL ist eine chronische Leukämie, die durch eine Ausdehnung des monoklonalen LGL-Phänotyps aktivierter zytotoxischer T-Lymphozyten oder seltener NK-Zellen gekennzeichnet ist.5 Das Durchschnittsalter der Diagnose beträgt 60 Jahre und wird häufig mit Autoimmunerkrankungen, insbesondere RA, in Verbindung gebracht, wurde aber auch bei Colitis ulcerosa, Sjögren-Syndrom, Lupus erythematodes und multipler Sklerose 1 beschrieben. Patienten mit RA, die mit LGLL assoziiert sind, haben ein ähnliches klinisches Erscheinungsbild wie FS. In der Regel handelt es sich um Patienten mit langjähriger RA, schweren Gelenkschäden und erheblichen Folgen sowie einer erhöhten Häufigkeit extraartikulärer Manifestationen wie rheumatoiden Knötchen, Lymphadenopathie, prätibialen Geschwüren, Pleuritis, Hautpigmentierung, Neuropathie oder Episkleritis.6 Der Patient hatte auch eine weitere seltene Komplikation der RA, der rheumatoiden Pneumokoniose oder des Caplan-Syndroms, die durch das Auftreten von Lungenknoten mit einer Histopathologie gekennzeichnet war, die der typischer rheumatoider Knoten bei Patienten mit beruflicher Exposition gegenüber anorganischen Stäuben in der Vorgeschichte ähnlich war wie Kieselsäure, Kohle oder Granit.7 Unseres Wissens ist der vorliegende Fall der erste, der die Darstellung des Caplan-Syndroms und der LGLL bei demselben Patienten beschreibt. In den meisten Fällen ist die Präsentation LGLL schwere Neutropenie mit rezidivierenden bakteriellen Infektionen assoziiert. Die am häufigsten beteiligten Mikroorganismen sind Staphylococcus aureus, Streptococcus spp. und gramnegative Bazillen. Weniger häufig Anämie, Fieber, Nachtschweiß und Leber- und Milzvergrößerung5 kann es auch begleiten. Bis zu einem Drittel der Patienten mit LLGG haben zum Zeitpunkt der Diagnose keine offensichtliche klinische RA-Aktivität, behalten jedoch hohe ESR-Werte bei.6 Bis zu 40% der Patienten mit FS haben eine LGL-Lymphozytose.7 Diese Tatsache, zusammen mit der klinischen Ähnlichkeit und Assoziation mit HLA-DR4, hat stark darauf hingewiesen, dass FS und LGLL, die mit RA assoziiert sind, Ausdrücke derselben Entität sind, die durch die Proliferation von LGL gekennzeichnet sind.8 Andere Formen umfassen auch mildere Formen wie reaktive Lymphozytose und Infektionen bis hin zu aggressiveren Formen von NK5 LGLL. Die LGLL-Diagnose basiert auf dem Befund einer monoklonalen Expansion von LGL im peripheren Blut und Knochenmark mit einem charakteristischen Immunphänotyp (CD3+, CD4−, CD8 +, CD16+, CD28− und CD57+). Die Klonalität wird durch Untersuchung des reTCR9-Gens bestätigt. Im Allgemeinen hat LGL eine chronische und indolente Progression mit einem mittleren Überleben von 10 Jahren.1 In seltenen Fällen, insbesondere wenn die Expansion auf LGL mit NK-Phänotyp zurückzuführen ist, kann sich diese Leukämie aggressiver verhalten.5 Die häufigste Indikation für eine Behandlung sind wiederkehrende Infektionen und seltener Anämie, symptomatische Splenomegalie oder das Auftreten schwerer B-Symptome B1.
Die Erstlinientherapie bei LGLL sind Immunsuppressiva allein, insbesondere Methotrexat (10 mg / Woche), Cyclosporin A (1-1,5 mg / kg / 2-mal täglich) oder orales Cyclophosphamid (50-100 mg / Tag). Diese Behandlung ist bei etwa 50% der Patienten wirksam und erreicht die Korrektur von Zytopenien, aber nicht die Ausrottung von Leukämiezellen.1 Glukokortikoide können verwendet werden, um die Reaktion zu beschleunigen, und Granulopoese-stimulierende Faktoren sind bei der anfänglichen Behandlung von Neutropenie nützlich. Bei refraktären Patienten und bei Patienten mit sehr aggressiver Präsentation wurde die Behandlung mit CHOP-ähnlichen Chemotherapien (Cyclophosphamid, Vincristin, Doxorubicin und Prednison) und anderen Lymphomschemata versucht, ihre Wirksamkeit jedoch nicht eindeutig nachgewiesen. Andere getestete Behandlungen sind Purinanaloga, Alemtuzumab, Bortezomib, Splenektomie und allogene Knochenmarktransplantation mit variablen Ergebnissen.3
Schlussfolgerungen
Sowohl FS als auch LGLL sind seltene Komplikationen der RA, die bei langjährigen Erkrankungen mit erheblichen strukturellen Schäden und extraartikulären Manifestationen auftreten. Bei Patienten mit lang anhaltender RA und Neutropenie sollte das Vorhandensein klonaler Proliferationen von LGL im peripheren Blut und / oder Knochenmark ausgeschlossen werden, um die Diagnose einer LGLL zu ermöglichen. Die Erstlinienbehandlung ist die Verwendung von Immunsuppressiva wie niedrig dosiertem Methotrexat, und Glukokortikoide können mit Granulopoese-stimulierenden Faktoren assoziiert sein. Andere Behandlungsmodalitäten wie Chemotherapie oder Splenektomie haben in einigen refraktären Fällen variable Ergebnisse gezeigt.
Ethische Angaben
Schutz von Menschen und Tieren. Die Autoren erklären, dass für diese Untersuchung keine Experimente an Menschen oder Tieren durchgeführt wurden.
Vertraulichkeit der Daten. Die Autoren erklären, dass sie die Protokolle ihres Arbeitszentrums zur Veröffentlichung von Patientendaten befolgt haben und dass alle in die Studie einbezogenen Patienten ausreichende Informationen erhalten und ihre schriftliche Einwilligung nach Aufklärung zur Teilnahme an dieser Studie erteilt haben.