Leukemie met grote granulaire lymfocyten als complicatie van reumatoïde artritis | Reumatología Clínica
introductie
Felty ‘ s syndrome (FS) is een zeldzame systemische complicatie (minder dan 1%) van reumatoïde artritis (RA), gekarakteriseerd door de triade van RA, persisterende neutropenie (3
) en splenomegalie van verschillende grootte die kan variëren van subklinische splenomegalie, alleen detecteerbaar door beeldvorming, tot massieve splenomegalie. Het komt voornamelijk voor in langdurige gevallen met ernstige gewrichtsaandoeningen en extraarticulaire manifestaties, en heeft een sterke associatie met het HLA-DR4 haplotype (bijna 95% van de gevallen).2 in 30% -40% van de patiënten met FS is er een expansie van grote granulaire lymfocyten (LGL).1 LGL vertegenwoordigt 10% -15% van circulerende mononucleaire cellen en morfologisch geà dentificeerd door hun grote grootte (15–18µm), hun ronde of ingesprongen kern en overvloedig cytoplasma met azurofiele korrels. Het fenotype van deze cellen kan cytotoxische T− lymfocyt (CD8+, CD57+) of natural killer (NK) (CD3−, CD8 -, CD56+) zijn.3 Wanneer de uitbreiding van LGL monoklonaal is en met infiltratie van het beendermerg en de milt door deze cellen wordt geassocieerd, wordt het grote granulaire lymfocyte leukemie (LGLL) genoemd en wordt beschouwd als een chronische low-grade lymfoproliferative wanorde. De klinische presentatie is vergelijkbaar met die van FS, waarbij de verhoogde gevoeligheid voor bacteriële infecties geassocieerd met neutropenie, bloedarmoede en splenomegalie wordt benadrukt, zoals ook “pseudo-Felty”wordt genoemd.3,4 klinische presentatie
een 70-jarige gepensioneerde werknemer uit een granietgroeve werd op 43-jarige leeftijd gediagnosticeerd met seropositieve RA met betrokkenheid van handen, voeten, knieën en heupen. Vervolgens ontwikkelde hij pneumoconiose en pulmonale reumatoïde knobbeltjes (Fig. 1), gediagnosticeerd als het syndroom van Caplan. Tijdens zijn progressie werd hij behandeld met NSAID ‘ s, corticosteroïden, goudzouten, cyclosporine en methotrexaat. Ondanks dit, ontwikkelde de patiënt structurele schade in de handen, voeten en heupen, die plaatsing van prothese in beide heupen op respectievelijk 51 en 54 jaar oud vereisen. In de afgelopen jaren bleef zijn ziekte stabiel, behandeld met methotrexaat 10 mg per week en lage doses glucocorticoïden, zonder bewijs van inflammatoire gewrichtsactiviteit. Hij presenteerde ‘zwanenhals’ misvormingen in alle vingers en reumatoïde knobbeltjes op de ellebogen. 12 maanden geleden kreeg hij plotseling koorts en pijn in zijn rechter lies, met daaropvolgende septische shock, waaruit een infectie van de rechter heup door Salmonella spp.bleek. Methotrexaat en glucocorticoïden werden opgeschort, en hij werd behandeld met langdurige antibiotische therapie en gedeeltelijke vervanging van de prothese. Laboratoriumtesten toonden aanhoudende neutropenie aan, ondanks het staken van myelotoxische geneesmiddelen en de verbetering van sepsis, die 0neutrofielen/mm3 bereikte. Retrospectief, bij het beoordelen van het aantal neutrofielen, had hij lage tellingen een jaar eerder, tussen 1800 en 1000/mm3. De rest van het bloedbeeld en de biochemie waren normaal. De ESR was 80mm / h en CRP 111mg / l. hij handhaafde hoge niveaus van reumatoïde factor (6930U / ml) en anti-CCP (300U/ml) en had ook polyclonale hypergammaglobulinemie. Antinucleaire antilichamen en extraheerbare nucleaire antigenen waren negatief en complementniveaus lagen binnen de normale waarden. HLA-typering toonde aan dat hij het haplotype DRB1*0404 (DR4) droeg en milde splenomegalie werd gedetecteerd (13,7 cm) op abdominale computertomografie.

pneumoconiose en pulmonale reumatoïde knobbeltjes.
het perifere bloeduitstrijkje vertoonde lymfocytose door LGL (Fig. 2), dat in het immunofenotype overeenkwam met 42% van het totaal aan leukocyten, met een afwijkend fenotype van cytotoxische T− lymfocyten (CD3+, CD8+, CD5+, CD7+/−, CD4−, CD56-en DR+). De beenmergbiopsie toonde 20% van de totale cellulariteit van het beenmerg, overeenkomend met dezelfde klonale expansie (bevestigd door herschikking van het variabele gebied van TCR-gamma). Op basis van deze bevindingen werd een diagnose van lgll gesteld en werd de behandeling met 15 mg methotrexaat per week hervat ondanks 3 maanden met aanhoudende neutropenie (3
), waardoor frequente toediening van granulopoëse-stimulatorische factoren noodzakelijk was. Vervolgens werd hij in hoge doses behandeld met cyclofosfamide, vincristine en prednison. Na 6 maanden behandeling houdt neutropenie aan.
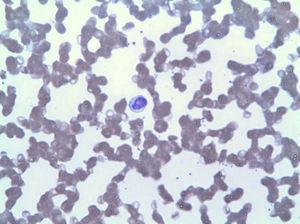
lymfocytose veroorzaakt door LGL.
discussie
LGLL is een chronische leukemie gekenmerkt door expansie van het LGL monoklonale fenotype van geactiveerde cytotoxische T-lymfocyten of minder vaak NK-cellen.De gemiddelde leeftijd van de diagnose is 60 jaar en het wordt vaak geassocieerd met auto-immuunziekten, met name RA, maar is ook beschreven in colitis ulcerosa, het syndroom van Sjögren, lupus erythematosus en multiple1 sclerose. Patiënten met RA die geassocieerd zijn met LGLL hebben een klinische presentatie die vergelijkbaar is met die van FS. Het zijn meestal patiënten met langdurige RA, ernstige gewrichtsschade en significante gevolgen, en verhoogde frequentie van extra-articulaire manifestaties zoals reumatoïde knobbeltjes, lymfadenopathie, pretibiële ulcera, pleuritis, huidpigmentatie, neuropathie of episcleritis.De patiënt had ook een andere zeldzame complicatie van RA, reumatoïde pneumoconiose of het syndroom van Caplan, gekenmerkt door het verschijnen van pulmonale knobbeltjes met histopathologie vergelijkbaar met die van typische reumatoïde knobbeltjes bij patiënten met een voorgeschiedenis van beroepsmatige blootstelling aan anorganisch stof zoals silica, steenkool of graniet.7 voor zover wij weten, is dit het eerste geval waarin het syndroom van Caplan en LGLL bij dezelfde patiënt worden beschreven. In de meeste gevallen is de presentatie lgll ernstige neutropenie geassocieerd met recidiverende bacteriële infecties. De meest betrokken micro-organismen zijn Staphylococcus aureus, Streptococcus spp. en gram-negatieve bacillen. Minder vaak bloedarmoede, koorts, nachtelijk zweten en lever en milt vergroting 5 kan ook gepaard gaan met het. Tot een derde van de patiënten met LLGG heeft geen duidelijke klinische Ra-activiteit op het moment van de diagnose, maar behoudt een hoog niveau van ESR.6 tot 40% van de patiënten met FS hebben LGL-lymfocytose.7 dit feit, samen met de klinische gelijkenis en associatie met HLA-DR4 heeft sterk gesuggereerd dat FS en LGLL geassocieerd met RA zijn uitdrukkingen van dezelfde entiteit gekenmerkt door de proliferatie van LGL.8 andere vormen omvatten ook mildere vormen zoals reactieve lymfocytose en besmettingen aan agressievere vormen van NK5 LGLL. De diagnose LGLL is gebaseerd op de bevinding van een monoklonale expansie van LGL in perifeer bloed en beenmerg met een karakteristiek immunofenotype (CD3+, CD4−, CD8 +, CD16+, CD28− en CD57+). De clonaliteit wordt bevestigd door het reTCR9 gen te bestuderen. Over het algemeen heeft LGL een chronische en indolente progressie, met een gemiddelde overleving van 10 jaar.1 in zeldzame gevallen, vooral wanneer de uitbreiding te wijten is aan LGL met NK fenotype, kan deze leukemie zich agressiever gedragen.5 de meest voorkomende indicatie voor de behandeling zijn terugkerende infecties en, minder vaak, bloedarmoede, symptomatische splenomegalie of het optreden van ernstige B-symptomen B1.
de eerstelijnsbehandeling bij LGLL zijn alleen immunosuppressieve geneesmiddelen, in het bijzonder methotrexaat (10 mg/week), cyclosporine A (1-1, 5 mg/kg/2 maal daags) of oraal cyclofosfamide (50-100 mg/dag). Deze behandeling is effectief in ongeveer 50% van de patiënten, het bereiken van de correctie van cytopenieën, maar niet het uitroeien van leukemische cellen.1 glucocorticoïden kunnen worden gebruikt om de respons te versnellen en granulopoëse stimulerende factoren zijn nuttig bij de initiële behandeling van neutropenie. Bij refractaire patiënten en bij patiënten met een zeer agressieve presentatiebehandeling met chemotherapieregimes vergelijkbaar met CHOP (cyclofosfamide, vincristine, doxorubicine en prednison) en andere schema ‘ s voor lymfoom zijn geprobeerd, maar hebben hun werkzaamheid niet duidelijk aangetoond. Andere geteste behandelingen zijn purine-analogen, Alemtuzumab, bortezomib, splenectomie en allogene beenmergtransplantatie, met wisselende resultaten.3
conclusies
zowel FS als LGLL zijn zeldzame complicaties van RA, die optreden bij langdurige ziekte, met significante structurele schade en extra-articulaire manifestaties. Bij patiënten met langdurige RA en neutropenie dient de aanwezigheid van clonale proliferaties van LGL in perifeer bloed en/of beenmerg te worden uitgesloten, zodat de diagnose van LGLL kan worden gesteld. Eerstelijnsbehandeling is het gebruik van immunosuppressieve geneesmiddelen, zoals een lage dosis methotrexaat, en glucocorticoïden kunnen worden geassocieerd met granulopoiese stimulerende factoren. Andere behandelingsmodaliteiten zoals chemotherapie of splenectomie hebben in sommige refractaire gevallen wisselende resultaten laten zien.
ethische informatie
bescherming van personen van mens en dier. De auteurs verklaren dat er voor dit onderzoek geen experimenten zijn uitgevoerd op mensen of dieren.
vertrouwelijkheid van gegevens. De auteurs verklaren dat zij de protocollen van hun werkcentrum voor de publicatie van patiëntgegevens hebben gevolgd en dat alle patiënten die in het onderzoek zijn opgenomen voldoende informatie hebben ontvangen en schriftelijk hun geïnformeerde toestemming hebben gegeven om aan dat onderzoek deel te nemen.