Wichtige Ankündigung
Amyloidose ist eine Gruppe von Krankheiten, die ein gemeinsames Merkmal der Ablagerung von Amyloidfibrillen in verschiedenen Organen und Geweben aufweisen. Die systemischen Amyloidose-Typen sind alle sehr unterschiedlich in Bezug auf die biochemische Natur der Amyloid-Ablagerungen und des Vorläufer-Amyloid-Proteins. Einige werden erworben und andere vererbt. Einige Arten teilen klinische Merkmale der Krankheit in Bezug auf Organbeteiligung und Dysfunktion. Es ist entscheidend, das Vorläufer-amyloidogene Protein zu definieren und Amyloidablagerungen genau zu typisieren, um eine genaue Behandlung anbieten zu können, die Prognose zu bewerten und bei Bedarf eine genetische Beratung vorzuschlagen.
AL-Amyloidose (Primär)
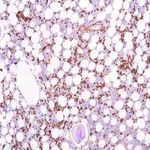
AL ( Immunglobulin-Leichtkette) Amyloidose ist eine erworbene Plasmazellstörung, bei der eine monoklonale Immunglobulin-Leichtkette im Knochenmark produziert wird und normalerweise im Blut oder Urin vorkommt. AL-Amyloidose tritt gelegentlich bei multiplem Myelom, Lymphom oder Waldenstorm-Makroglobulinämie auf. Die Amyloidfibrillen bei dieser Art von Amyloidose bestehen aus Immunglobulin-Leichtkettenproteinen (Kappa oder Lambda). Symptome können in jedem Organ des Körpers auftreten und umfassen Herzinsuffizienz, Protein im Urin oder Nierenversagen, vergrößerte Leber, Neuropathie oder vergrößerte Zunge. Die Behandlung mit einer Chemotherapie, einschließlich einer hochdosierten Chemotherapie mit Stammzelltransplantation und anderen Wirkstoffen, kann die Knochenmarkerkrankung in Remission versetzen, was zu einer Verbesserung der Organfunktionsstörung führt und die Zielorgane schützt. Dies ist eine der häufigsten Arten von Amyloidose in den Vereinigten Staaten diagnostiziert.
AA-Amyloidose (sekundär)
AA-Amyloidose wird durch eine chronische Infektion oder eine entzündliche Erkrankung wie rheumatoide Arthritis, familiäres Mittelmeerfieber (FMF), Osteomyelitis oder granulomatöse Ileitis verursacht. Eine Infektion oder Entzündung verursacht eine Erhöhung eines Akutphasenproteins, SAA, von dem sich ein Teil als Amyloidfibrillen ablagert. Daher wird es als AA-Amyloidose bezeichnet. AA Amyloidose beginnt in der Regel als Krankheit in den Nieren, aber auch andere Organe können betroffen sein. Eine medizinische oder chirurgische Behandlung der zugrunde liegenden chronischen Infektion oder entzündlichen Erkrankung kann das Fortschreiten dieser Art von Amyloidose verlangsamen oder stoppen.
ATTR amyloidosis
ATTR amyloidosis (ATTRm or ATTRwt)
There are several types of inherited amyloidoses, the most common of which is caused by a mutation in the transthyretin (TTR) gene that produces abnormal transthyretin protein. The abnormal TTR protein deposits as amyloid fibrils: Hence, it is termed ATTR amyloidosis. Krankheitssymptome sind in der Regel Neuropathie und Kardiomyopathie und treten im mittleren bis späten Leben auf. ATTR-Amyloidose tritt in Familien mit fast jedem ethnischen Hintergrund auf. Mehr als 100 verschiedene Mutationen in Transthyretin sind bekannt und die meisten verursachen Amyloidose. ATTR kann auch mit dem unmutierten Wildtyp-TTR-Protein auftreten, das normalerweise bei älteren Männern eine Kardiomyopathie verursacht; Dies wurde früher als „senile systemische Amyloidose“ bezeichnet, ist aber jetzt als ATTRwt-Amyloidose bekannt. Zu den Behandlungsmöglichkeiten gehören TTR-Stabilisatoren (Diflunisal oder Tafamidis) oder Gen-Schalldämpfer oder TTR-Produktion oder Lebertransplantation.
Andere erbliche Amyloidosen
Es gibt andere Genmutationen, die Proteine produzieren, die Amyloidose verursachen. Diese sind sehr selten. Dazu gehören Apolipoprotein A-I (AApoAI), Apolipoprotein A-II (AApoAII), Gelsolin (AGel), Fibrinogen (AFib) und Lysozym (ALys). Ein Protein, das als Lect2 bekannt ist, kann auch Amyloidose verursachen; Es ist nicht klar, ob dies vererbt wird oder nicht.
Beta-2-Mikroglobulinamyloidose (Abeta2m)
Die Beta-2-Mikroglobulinamyloidose wird durch chronisches Nierenversagen verursacht und tritt häufig bei Patienten auf, die seit vielen Jahren dialysiert werden. Amyloidablagerungen bestehen aus dem Beta-2-Mikroglobulinprotein, das sich in Geweben, insbesondere in der Nähe von Gelenken, ansammelt, wenn es aufgrund von Nierenversagen nicht von der Niere ausgeschieden werden kann.
Lokalisierte Amyloidose (ALoc)
Es gibt viele Arten von lokalisierten Amyloidosen. Lokalisierte Amyloidablagerungen in den Atemwegen (Luftröhre oder Bronchus), im Auge oder in der Harnblase werden häufig durch die lokale Produktion von Immunglobulin-Leichtketten verursacht, die nicht aus dem Knochenmark stammen. Lokalisierte AL kann mit Strahlentherapie behandelt werden, und es und andere lokalisierte Formen sind manchmal einer Operation zugänglich. Andere lokalisierte Arten von Amyloidose sind mit endokrinen Proteinen oder Proteinen assoziiert, die in der Haut, im Herzen und an anderen Stellen produziert werden. Diese werden normalerweise nicht systemisch.