Idiopathische Livedo Racemosa mit Splenomegalie und diffuser Lymphadenopathie
Das Sneddon-Syndrom (SS) wurde erstmals 1965 bei Patienten mit persistierender Livedo Racemosa und neurologischen Ereignissen beschrieben.1 Da die anderen Manifestationen von SS unspezifisch sind (z. B. Hypertonie, Herzklappenerkrankung, arterieller und venöser Verschluss), verzögert sich die Diagnose häufig. Viele Patienten, bei denen prodromale neurologische Symptome wie Kopfschmerzen, Depressionen, Angstzustände, Schwindel und Neuropathie auftreten, werden häufig vor der Entwicklung ischämischer Hirnmanifestationen einem Arzt vorgelegt2, erhalten jedoch selten die richtige Diagnose. Der Beginn zerebraler okklusiver Ereignisse tritt typischerweise bei Patienten unter 45 Jahren auf und kann als vorübergehende ischämische Attacke, Schlaganfall oder intrakranielle Blutung auftreten.3 Die Krankheit ist bei Frauen häufiger als bei Männern (Verhältnis 2:1). Die genaue Pathogenese von SS ist noch unbekannt, und obwohl es als eine separate Einheit von systemischem Lupus erythematodes und anderen Antiphospholipid-Erkrankungen gedacht wurde, wurde postuliert, dass eine immunologische Dysfunktion Gefäßwände schädigt, die zu Thrombosen führen.
Hautbefunde im Zusammenhang mit SS betreffen kleine bis mittelgroße dermal-subdermale Arterien. Die Histopathologie bei einigen Patienten zeigt eine Proliferation des Endothels und Fibrinablagerungen mit anschließender Obliteration der beteiligten Arterien.4 Bei vielen Patienten, einschließlich unseres Patienten, zeigt die histopathologische Untersuchung der betroffenen Haut keine spezifischen Anomalien.1 Zelger et al5 berichteten über die Abfolge histopathologischer Hautereignisse bei einer Reihe von Antiphospholipid-negativen SS-Patienten. Die Autoren berichteten, dass nur kleine Arterien an der Dermis-Subcutis-Verbindung beteiligt waren und ein Fortschreiten der endothelialen Dysfunktion beobachtet wurde. Die Autoren glaubten, dass es mehrere unspezifische Stadien vor dem Fibrinverschluss der beteiligten Arterien gab.5 Stadium I beinhaltete die Lockerung von Endothelzellen mit unspezifischer perivaskulärer lymphozytärer Infiltration mit perivaskulärer Entzündung und lymphozytärer Infiltration, die den Hauptantrieb der Krankheit darstellten.5,6 Es wird angenommen, dass dieses Stadium nur von kurzer Dauer ist, weshalb es bei SS-Patienten viele Jahre lang unentdeckt blieb. Die Stadien II bis IV verlaufen durch Fibrinablagerung und Okklusion.5 Histologische Merkmale der Stadien I bis II wurden aufgrund der späten Diagnose von SS nicht berichtet. Patienten im Stadium I weisen typischerweise eine durchschnittliche Symptomdauer von 6 Monaten mit wenigen neurologischen Symptomen auf, am häufigsten sind Parästhesien der Beine.5
Fallbericht
Eine 37-jährige Frau mit epigastrischer Empfindlichkeit auf der linken Seite und Splenomegalie, die auf der Computertomographie gesehen wurde, wurde von einem Hämatologen zur Beurteilung eines retikulären Hautausschlags auf der linken Seite der Flanke von 9 Monaten Dauer mit einer vermuteten Diagnose einer fokalen Melanodermie überwiesen. Ihre Krankengeschichte war bemerkenswert für einen angeborenen Ventrikelseptumdefekt und eine Aortenisthmusstenose, sowie Endometriose, Myalgie, und Gelenksteifigkeit, die sich alle im letzten Jahr entwickelt hatten. Ihre Krankengeschichte war auch bemerkenswert für Nephrolithiasis, Reizdarmsyndrom und chronische Sinusitis sowie psychiatrische Depressionen und Angststörungen. Vor kurzem wurde bei ihr eine mittelschwere Hypertonie diagnostiziert und sie hatte in den letzten Jahren Schwierigkeiten, schwanger zu werden, mit 3 aufeinanderfolgenden Fehlgeburten im ersten Trimester. Neurologische Symptome waren Neuropathie mit den Füßen, intermittierende Parästhesien der Beine und eine Geschichte von chronischen Migräne-Kopfschmerzen für mehrere Monate.Die dermatologische Untersuchung ergab eine leicht übergewichtige Frau mit einem 25 × 30 cm großen dunklen, erythematösen, unregelmäßigen, netzartigen Muster auf der linken Seite des oberen und unteren Rumpfes (Abbildung 1). Die Livedo racemosa wurde durch Temperaturänderungen nicht verändert und war seit mehr als 9 Monaten unverändert. Es gab keine Anzeichen von Pruritus oder Ulzerationen, und Bereiche von Livedo racemosa waren leicht palpationsempfindlich.
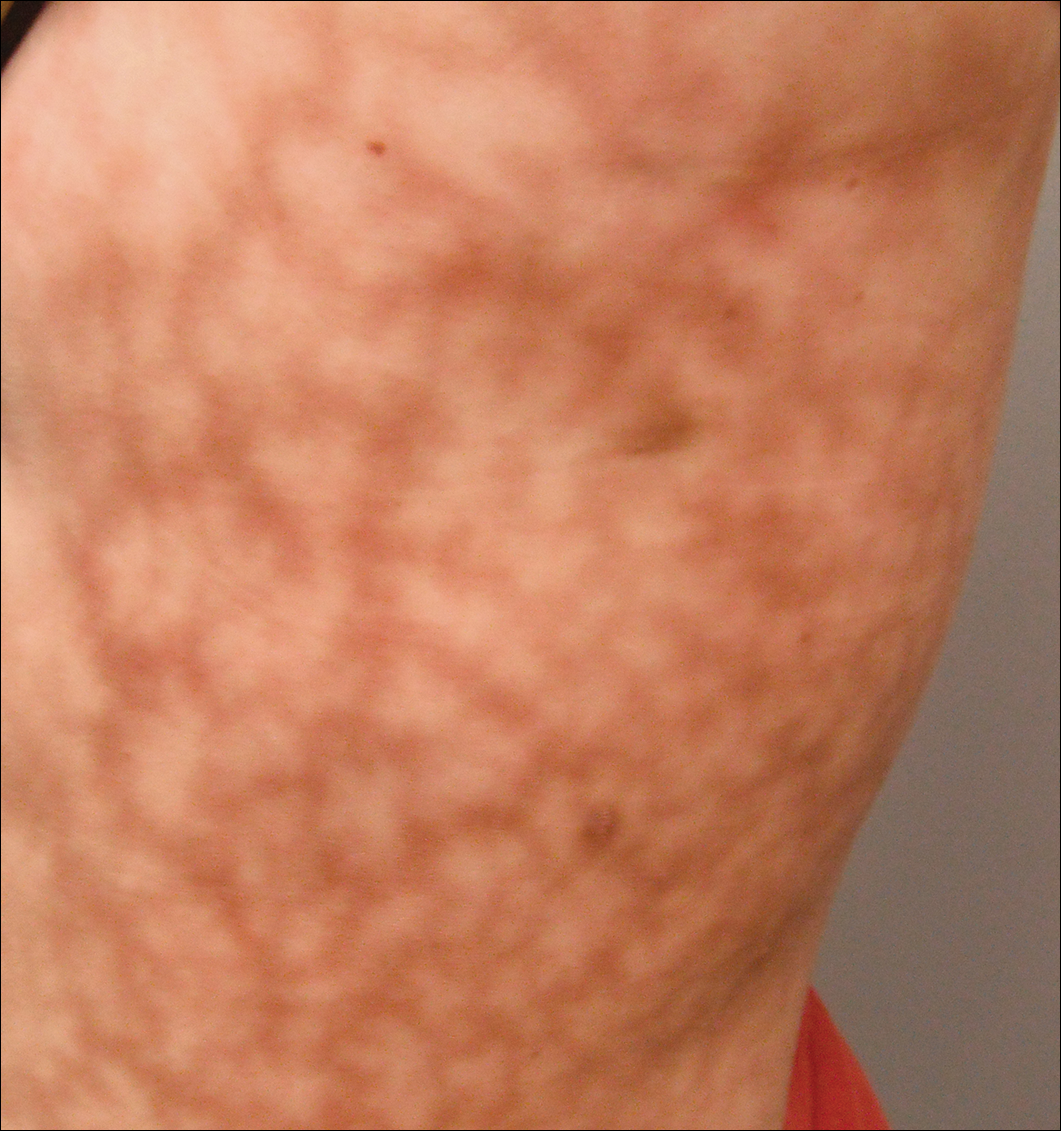
Abbildung 1. Livedo racemosa präsentiert sich als netzartiges violettes Muster auf der linken Seite des Stammes mit den Maßen 25 × 30 cm.
Wir führten 2 Sätze von drei 4-mm-Biopsien durch. Der erste Satz zielte auf Bereiche innerhalb des violetten Musters ab, während der zweite Satz auf Bereiche normalen Gewebes zwischen den gesprenkelten Bereichen abzielte. Alle 6 Proben zeigten ein oberflächliches perivaskuläres lymphozytisches Infiltrat ohne Anzeichen einer Vaskulitis oder Bindegewebserkrankung. Die Gefäße zeigten keine Mikrothromben oder umgebende Fibrose. Innerhalb der Epidermis wurden keine Eosinophilen identifiziert. Es gab keine Hinweise auf ein erhöhtes dermales Mucin. Sowohl der oberflächliche als auch der tiefe Gefäßplexus waren unauffällig und zeigten keine Anzeichen von Schäden an den Wänden (Abbildung 2).
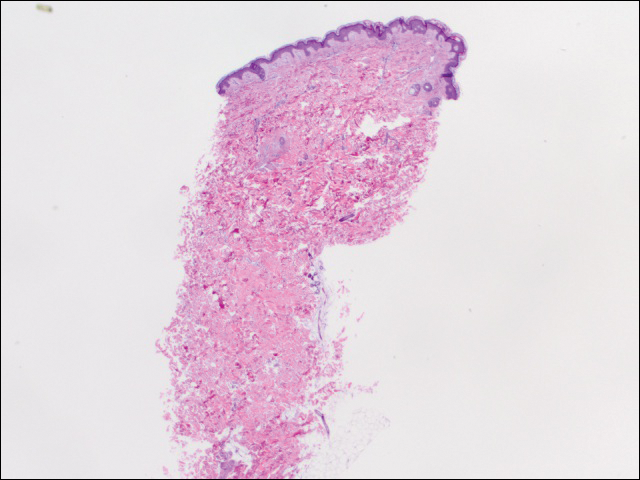
Abbildung 2. Die Stanzbiopsie von der linken Seite des Rumpfes zeigte eine fokale Melanodermie und ein spärliches oberflächliches perivaskuläres lymphozytisches Infiltrat ohne Anzeichen von Vaskulitis, Mikrothromben oder Fibrinablagerung (H&E, ursprüngliche Vergrößerung ×20).
Um andere mögliche Ursachen von Livedo racemosa auszuschließen, wurden eine vollständige Blutzellzahl, ein umfassendes metabolisches Panel, ein Gerinnungsprofil, ein Lipasetest, eine Urinanalyse, serologische Tests und eine immunologische Aufarbeitung durchgeführt. Die Lipase lag im Referenzbereich. Die vollständige Blutzellzahl ergab eine leichte Anämie, während der Rest der Werte innerhalb des Referenzbereichs lag. Eine immunologische Aufarbeitung umfasste Sjögren-Syndrom-Antigen A, Sjögren-Syndrom-Antigen B, Anticardiolipin-Antikörper und antinukleäre Antikörper, die alle negativ waren. Die Familiengeschichte war bemerkenswert für Verwandte ersten Grades mit systemischem Lupus erythematodes und Morbus Crohn.
Die Computertomographie ergab eine Vergrößerung der Milz sowie eine Periaorten-, Portacaval- und Porta hepatis-Lymphadenopathie. Basierend auf den Laborbefunden und dem klinischen Erscheinungsbild sowie der Krankengeschichte des Patienten lautete die Ausschlussdiagnose idiopathische Livedo racemosa mit unbekanntem Fortschreiten zur ausgewachsenen SS. Die Patientin erfüllte nicht die aktuellen diagnostischen Kriterien für SS, und ihre immunologischen Studien konnten keine vorhandenen Antikörper bestätigen, aber die Beteiligung des retikuloendothelialen Systems wies auf die Produktion von Antikörpern hin, die bei Labortests noch nicht nachweisbar waren.